


Co znajdziesz w artykule?
- Rola witaminy B6 w organizmie
- Przyczyny uwarunkowanych genetycznie drgawek pirydoksynozależnych
- Charakterystyka chorób uwarunkowanych genetycznie, które prowadzą do drgawek zależnych od pirydoksyny, tj. zazwyczaj ujawniających się w okresie noworodkowym, ustępujących po podaży leczniczych dawek witaminy B6 i opornych na klasyczne leki przeciwpadaczkowe
Spis treści
- Do czego jest potrzebna witamina B6
- Deficyt dehydrogenazy semialdehydu α-aminoadypinowego
- Deficyt oksydazy fosforanu pirydoksaminy
- Deficyt białka wiążącego fosforan pirydoksalu
- Hiperprolinemia typu II (deficyt dehydrogenazy prolino-5-karboksylanu)
- Wrodzona hipofosfatazja i hiperfosfatazja
- Podsumowanie
Witamina B6 jest kluczowym kofaktorem niezbędnym do funkcjonowania wielu enzymów i szlaków metabolicznych w ludzkim organizmie. Jej niedobór, wynikający z wrodzonych wad metabolizmu, prowadzi do encefalopatii padaczkowej, ujawniającej się często od okresu noworodkowego i wczesnoniemowlęcego. Z reguły napady te są oporne na klasyczne leki przeciwpadaczkowe, ale ustępują po podaży terapeutycznej dawki witaminy B6 lub jej aktywnej formy – fosforanu pirydoksalu. W każdym przypadku drgawek
opornych na leczenie, szczególnie tych o nieustalonej etiologii, należy rozważyć wrodzoną wadę metabolizmu i podjąć próbę leczenia witaminą B6.
Do czego jest potrzebna witamina B6
Witamina B6 jest witaminą rozpuszczalną w wodzie, występuje w różnych postaciach: jako pirydoksyna, pirydoksal i pirydoksamina. Wszystkie te formy przekształcane są do aktywnej postaci – 5-fosforanu pirydoksalu (PLP – pyridoxal phosphate). Dzienne zapotrzebowanie na witaminę B6 u zdrowych niemowląt wynosi 0,1-0,3 mg i wzrasta do 1,2-1,4 mg/24 h u dorosłych. Pokarm kobiecy, produkty mleczne, zbożowe i mięso są bogate w witaminę B6 i obecnie rzadko obserwuje się jej niedobór wynikający z niedostatecznej podaży 1, 2 .
PLP odgrywa kluczową rolę w wielu reakcjach metabolicznych, szacuje się, że jako kofaktor jest niezbędny w ok. 140 reakcjach enzymatycznych, z czego 70 występuje w organizmie ludzkim. Reakcje te obejmują różne szlaki metaboliczne, w tym: szlaki przemiany aminokwasów i neuroprzekaźników, metabolizm kwasu foliowego, syntezę białek, węglowodanów i lipidów. Wpływa również na funkcję mitochondriów i erytropoezę. Poza rolą kofaktora enzymów witamina B6 działa w komórce również jako przeciwutleniacz, modyfikuje ekspresję i działanie receptorów dla hormonów steroidowych 3 .
Niedobór witaminy B6 może wynikać z różnych przyczyn, m.in. występuje u osób:
- z przewlekłą niewydolnością nerek (szczególnie w trakcie dializoterapii)
- z niewydolnością wątroby
- z reumatoidalnym zapaleniem stawów
- z cukrzycą typu 1.
Niedobór ten może też występować w czasie ciąży. Dodatkowo dostępność witaminy B6 może być zaburzona podczas leczenia glikokortykosteroidami, preparatami przeciwpadaczkowymi, izoniazydem czy cykloseryną (niedostępną w Polsce).
Niedobór witaminy B6 klinicznie objawia się w postaci łojotokowego zapalenia skóry, atrofii błony śluzowej języka z nadżerkami, zapalenia kącików ust, splątania czy neuropatii, dodatkowo opisano pojedyncze przypadki dorosłych, u których wystąpiły drgawki 2 .
Ze względu na istotną rolę PLP w metabolizmie neuroprzekaźników, zwłaszcza w syntezie przekaźnika hamującego – kwasu γ-aminomasłowego (GABA) – nie zaskakuje fakt, że różne wrodzone wady metabolizmu, prowadzące do niedoboru PLP, objawiają się padaczką niereagującą na klasyczne leki przeciwpadaczkowe i zwykle ustępują po podaży terapeutycznych dawek witaminy B6 1, 2, 3 .
Dotąd poznano 6 chorób uwarunkowanych genetycznie, które prowadzą do drgawek zależnych od pirydoksyny, tzn. drgawek, które zazwyczaj ujawniają się w okresie noworodkowym, ustępują po podaży leczniczych dawek witaminy B6 i są oporne na klasyczne leki przeciwpadaczkowe 1, 3 . Krótką charakterystykę ww. chorób przedstawiono w tabeli 1.
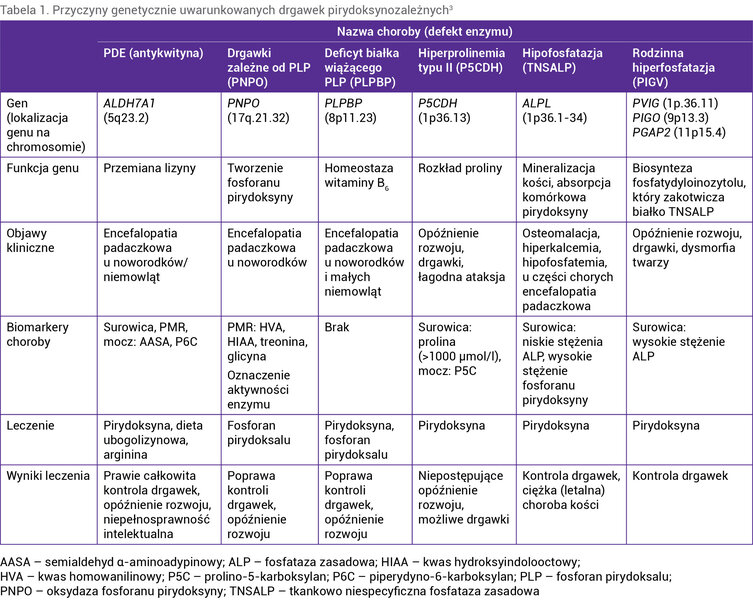
Tabela 1. Przyczyny genetycznie uwarunkowanych drgawek pirydoksynozależnych3
Deficyt dehydrogenazy semialdehydu α-aminoadypinowego
Dehydrogenaza semialdehydu α-aminoadypinowego (antykwityna, ALDH7A1) jest enzymem biorącym udział w metabolizmie lizyny (aminokwasu egzogennego) kodowanym przez gen ALDH7A1. Jej niedobór powoduje gromadzenie się w organizmie m.in. semialdehydu α-aminoadypinowego (AASA), kwasu pipekolowego i Δ1-piperydyno-6-karboksylanu (Δ 1 P6C) – ta ostatnia substancja wiąże i unieczynnia aktywną formę witaminy B6 (PLP). Jest to najczęstsza przyczyna drgawek pirydoksynozależnych (PDE – pyridoxine-dependent epilepsy) (OMIN#266100) 3, 4 .
Niedobór antykwityny dziedziczy się w sposób autosomalny recesywny; dotąd poznano 165 patogennych wariantów odpowiadających za chorobę. Częstość występowania szacowana jest na 1:60 000-1:250 000 żywych urodzeń (podobna częstość jak w przypadku deficytu biotynidazy czy galaktozemii) 5 .
W obrazie klinicznym PDE-ALDH7A1 dominuje encefalopatia padaczkowa. Drgawki nie reagują na leki przeciwpadaczkowe, ustępują natomiast po podaży pirydoksyny 6 . W klasycznej postaci napady pojawiają się od pierwszych godzin/dni życia dziecka, ale u 25-30% pacjentów pierwszy napad wystąpił po okresie noworodkowym. Najstarszy opisany pacjent z potwierdzoną mutacją w genie ALDH7A1 miał 17 lat w chwili wystąpienia drgawek. U części dzieci napady ujawniają się już w okresie prenatalnym; są odczuwane przez ciężarne jako rytmiczne ruchy lub czkawka płodu. U większości pacjentów napady mają charakter ogniskowy, ale wystąpić może każdy ich rodzaj: m.in. mioklonie, napady toniczno-kloniczne, napady skłonów. Większość pacjentów przed włączeniem leczenia miała w wywiadzie stany padaczkowe 7, 8, 9 .
Dzieci rodzą się zazwyczaj zdrowe, z donoszonej ciąży, mogą wystąpić trudności w adaptacji po urodzeniu, niższa może też być punktacja w skali Apgar. W momencie ujawnienia się drgawek większość pacjentów ma zaburzenia napięcia mięśniowego (hipotonia lub hipertonia), mogą występować drżenia i dystonie, rozdrażnienie, encefalopatia i nadmierna senność. Wśród ogólnych objawów najczęściej opisywane są zaburzenia karmienia, wymioty i zaburzenia oddychania 7, 10, 11, 12 .
Nie ma stałego wzorca zapisu elektroencefalograficznego (EEG) charakterystycznego dla tego rodzaju padaczki – zazwyczaj są to zmiany wieloogniskowe, ale również może być zapis ze zmianami uogólnionymi czy nawet typu cisza–wyładowanie. W pojedynczych przypadkach na początku choroby zapis EEG był prawidłowy. W wyniku neuroobrazowania ośrodkowego układu nerwowego (OUN) może nie być żadnych zmian, czasami stwierdza się dysplazję lub hipoplazję ciała modzelowatego, wentrikulomegalię, nieprawidłowości w istocie białej, krwawienie do OUN, zmiany zanikowe, szczególnie w obszarze kory mózgu, czy ogniskową dysplazję korową 7, 10, 11, 12 .
W podstawowych badaniach laboratoryjnych nie ma specjalnych odchyleń, czasami stwierdza się hipoglikemię i nieznacznie podwyższone stężenie kwasu mlekowego. Możliwe są zaburzenia w aminoacydogramie: w surowicy podwyższone stężenia treoniny, glicyny, tauryny i histydyny, a w płynie mózgowo-rdzeniowym (PMR) – poza ww. aminokwasami – również podwyższone stężenie glutaminianu, a obniżone PLP i GABA 7, 10, 11, 12 .
Markerem biochemicznym choroby są metabolity, które powstają na drodze przemiany lizyny: Δ 1 P6C, AASA i kwas pipekolowy. Stężenia Δ 1 P6C i AASA pozostają podwyższone również po włączeniu leczenia witaminą B6, chociaż w mniejszym stopniu niż przed leczeniem. Stężenie kwasu pipekolowego może się unormować w trakcie leczenia, poza tym może być niespecyficznie podwyższone w niewydolności wątroby, a także w chorobach peroksysomalnych, z kolei stężenie AASA może być również podwyższone w deficycie kofaktora molibdenu. Niedawno zidentyfikowano nowy biomarker 6-oxo-pipekolan (6-oxo-PIP), który jest możliwy do wykrycia również z suchej kropli krwi na bibule (jest termostabilny) lub w porcji moczu, co stwarza szansę na populacyjne badania skriningowe w kierunku tej choroby w przyszłości 13, 14 .
Obecnie zaleca się, aby przy podejrzeniu niedoboru antykwityny wykonać oznaczenie AASA i/lub Δ 1 P6C w surowicy, PMR lub moczu. Potwierdzenie choroby można również uzyskać na podstawie badania molekularnego 9 .
Postępowaniem z wyboru w przypadku stwierdzenia drgawek opornych na klasyczne leki przeciwpadaczkowe, szczególnie u noworodków i małych niemowląt, jest podaż witaminy B6. Pierwszą dawkę można podać dożylnie (100 mg), pamiętając, że u pacjentów z PDE-ALDH7A1 może dojść do bezdechu w trakcie podawania leku, dlatego należy taką próbę podejmować na oddziale intensywnej terapii z dostępem do respiratora. Następnie, niezależnie od odpowiedzi na podaż dożylną, należy kontynuować podaż witaminy B6 doustnie w dawce 30 mg/kg m.c./24 h w 3-4 dawkach podzielonych (maksymalnie 100-200 mg/24 h u noworodków i maksymalnie 300 mg/24 h u niemowląt). U dorosłych nie należy przekraczać dawki 500 mg/24 h (wyższe dawki mogą wywołać neuropatię). Są pacjenci, którzy odpowiadają na mniejsze dawki witaminy B6. Podawanie leku należy kontynuować do czasu wykluczenia biochemicznego i/lub molekularnego PDE-ALDH7A1, gdyż nie wszyscy chorzy odpowiadają na leczenie po pierwszych dawkach czy nawet dniach terapii. Zalecenia dotyczące leczenia PDE-ALDH7A1 wg rekomendacji z 2021 r. przedstawiono w tabeli 2 9 .
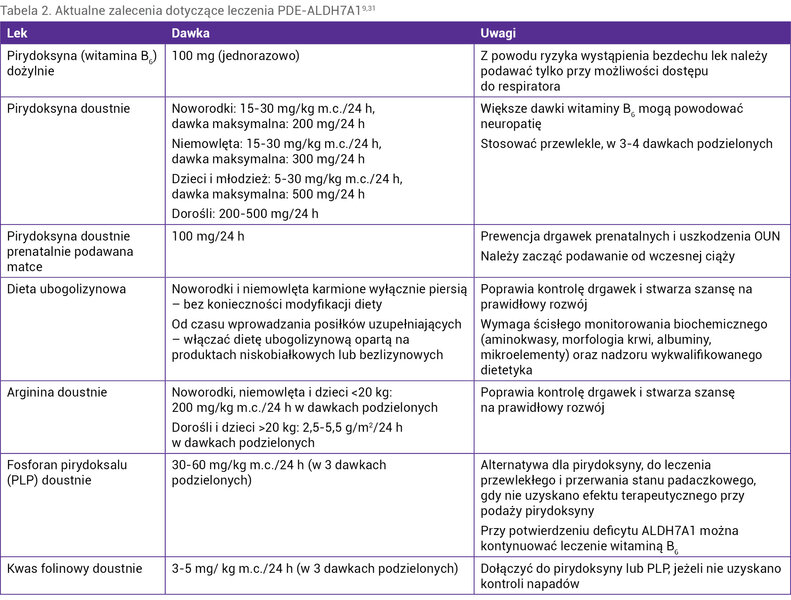
Tabela 2. Aktualne zalecenia dotyczące leczenia PDE-ALDH7A19,31
Jeżeli uda się uzyskać kontrolę drgawek po podaży witaminy B6, a nie potwierdzi się deficyt antykwityny, w diagnostyce różnicowej należy uwzględnić inne choroby zależne od witaminy B6 (tab. 1). W przypadku braku odpowiedzi na podaż witaminy B6 należy rozważyć inne wrodzone wady metabolizmu przebiegające z uporczywymi napadami, szczególnie te, które można leczyć lub ograniczyć ich postęp i objawy kliniczne specyficznym postępowaniem (tab. 3 i 4) 15, 16, 17 .
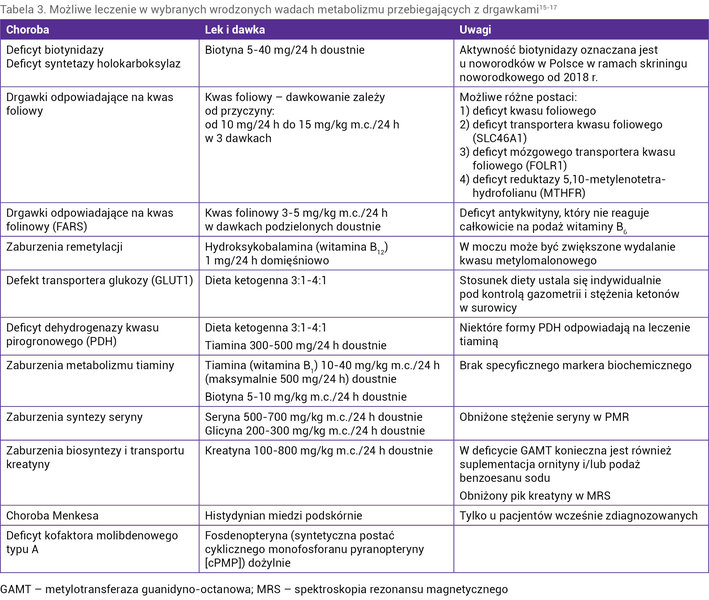
Tabela 3. Możliwe leczenie w wybranych wrodzonych wadach metabolizmu przebiegających z drgawkami15-17
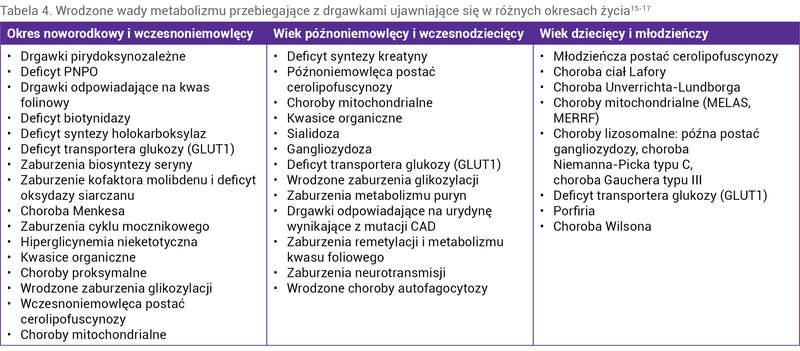
Tabela 4. Wrodzone wady metabolizmu przebiegające z drgawkami ujawniające się w różnych okresach życia15-17
U 90% pacjentów z PDE-ALDH7A1 udaje się uzyskać kontrolę napadów po podaży samej witaminy B6, jednak mimo to u 75% chorych stwierdza się opóźnienie rozwoju lub niepełnosprawność intelektualną w różnym stopniu. Okazuje się, że ograniczenie podaży lizyny, a tym samym obniżenie stężenia w płynach ustrojowych m.in. AASA czy Δ 1 P6C, stwarza możliwość poprawy rozwoju poznawczego w tej grupie pacjentów. Podobnie działa suplementacja argininy, aminokwasu, który z lizyną konkuruje o transporter w jelitach i przy przechodzeniu przez barierę krew–mózg w OUN 18, 19, 20, 21, 22, 23 . Pokarm kobiecy zawiera niewiele lizyny, dlatego noworodki i małe niemowlęta mogą być karmione mlekiem matki, a w okresie rozszerzania diety zaleca się produkty niskobiałkowe i preparaty bez lizyny (produkty spożywcze specjalnego przeznaczenia). U starszych dzieci i dorosłych w przypadku braku dostępności produktów ubogo- lub bezlizynowych można stosować dietę niskobiałkową. Należy jednak pamiętać, że dieta nie może być deficytowa i trzeba ją prowadzić pod kontrolą wykwalifikowanego dietetyka, z regularną kontrolą kliniczną i biochemiczną. Stężenie lizyny w surowicy powinno być utrzymywane w dolnych granicach wartości referencyjnej dla wieku 9 .
Suplementacja argininy u pacjentów z PDE-ALDH7A1 powinna wynosić 200 mg/kg m.c./24 h w dawkach podzielonych, dawka maksymalna u dorosłego to 4-5,5 g/m 2 /24 h 9 .
Opisano pojedyncze przypadki chorych z potwierdzonym biochemicznie i molekularnie deficytem antykwityny, którzy nie odpowiedzieli na terapeutyczne dawki witaminy B6, natomiast drgawki ustąpiły u nich po podaży kwasu folinowego. U chorych tych w chromatografii PMR stwierdzono niezidentyfikowany dotychczas pik substancji X. Dlatego przy uporczywych drgawkach u noworodków i małych dzieci, które nie odpowiedziały na leczenie witaminą B6, należy dołączyć kwas folinowy w dawce 3-5 mg/kg m.c./24 h w 2 dawkach podzielonych 3 .
Deficyt oksydazy fosforanu pirydoksaminy
Oksydaza fosforanu pirydoksaminy (PNPO – pyridoxamine phosphate oxidase) jest enzymem niezbędnym do powstania aktywnej formy witaminy B6, czyli PLP. Enzym ten kodowany jest przez gen PNPO (opisano 28 patogennych wariantów), a choroba dziedziczy się autosomalnie recesywnie. Jego deficyt (OMIN#6032870) prowadzi do encefalopatii padaczkowej, ujawniającej się od pierwszych dni życia 24 .
Obraz kliniczny jest bardzo podobny do obrazu występującego w przypadku deficytu antykwityny, uważa się jednak, że przebieg kliniczny jest cięższy niż w PDE-ALDH7A1. Dodatkowo ok. 40% pacjentów odpowiada redukcją lub ustąpieniem napadów po podaży samej witaminy B6. W badaniach biochemicznych może wystąpić hipoglikemia, kwasica metaboliczna i podwyższone stężenie kwasu mlekowego oraz treoniny i glicyny. W moczu stwierdza się wydalanie kwasu wanilinomlekowego (VLA – vanillin lactic acid) i kwasu homowanilinowego (HVA – homovanillic acid), w PMR zaś obniżone jest stężenia kwasu hydroksyindolooctowego (HIAA – hydroxyindoleacetic acid), HVA, pirydoksyny i PLP oraz argininy. Nie ma specyficznego biomarkera choroby, a obniżenie stężenie PLP w PMR zawsze powinno sugerować drgawki pirydoksynozależne. Potwierdzeniem choroby jest oznaczenie aktywności enzymu PNPO w suchej kropli krwi 3 .
Szybkie włączenie leczenia fosforanem pirydoksalu (3-60 mg/kg m.c./24 h) stwarza szansę na prawidłowy rozwój dziecka 25 . Dotychczas opisano w piśmiennictwie nieco ponad 60 pacjentów z potwierdzonym deficytem PNPO 3 .
Deficyt białka wiążącego fosforan pirydoksalu
To stosunkowo nowa choroba, pierwsze doniesienie o tej jednostce chorobowej zostało opublikowane w 2016 r. 26 , dotychczas opisano 27 pacjentów 7, 27 . Białko wiążące fosforan pirydoksalu (PLPBP – pyridoxal phosphate binding protein) kodowane jest przez gen PLPBP (wcześniej określany jako PROSC), choroba dziedziczy się w sposób autosomalny recesywny. Rola tego białka nie jest dokładnie poznana, wiadomo, że uczestniczy ono w homeostazie witaminy B6, prawdopodobnie zlokalizowane jest w mitochondriach i odgrywa rolę w metabolizmie mitochondriów.
W obrazie klinicznym dominuje encefalopatia padaczkowa, drgawki występują już prenatalnie lub ujawniają się w pierwszych dniach życia, często prowadząc do stanu padaczkowego i wczesnego zgonu w okresie niemowlęcym. U części pacjentów stwierdza się po urodzeniu małogłowie. Możliwe też są objawy ogólne, takie jak niedokrwistość, zaburzenia elektrolitowe czy biegunka. U chorych występują różnego stopnia zaburzenia rozwoju i niepełnosprawność intelektualna 26, 27 .
W PMR u pacjentów z deficytem PLPBP stwierdza się obniżone stężenie PLP, a w wyhodowanych fibroblastach – kumulację PLP. Dodatkowo występują objawy obniżonej aktywności enzymów zależnych od witaminy B6. Drgawki w znaczący sposób odpowiadają na leczenie witaminą B6 lub jej aktywną formą – PLP 7, 27 .
Hiperprolinemia typu II (deficyt dehydrogenazy prolino-5-karboksylanu)
Dehydrogenaza prolino-5-karboksylanu, kodowana przez gen ALDH4A1, bierze udział w przemianie proliny. Zahamowanie tej reakcji prowadzi do gromadzenia się wysokich stężeń proliny (w surowicy >1000 μmol/l) i prolino-5-karboksylanu (P5C), który podobnie jak Δ 1 P6C (w deficycie antykwityny) wiąże PLP. Choroba ujawnia się zazwyczaj w okresie noworodkowym i wczesnym niemowlęcym, pacjenci mają napady padaczkowe, a w późniejszym okresie występują także zaburzenie rozwoju, niepełnosprawność intelektualna oraz zaburzenia zachowania czy zaburzenia psychiczne (schizofrenia) 28, 29 .
Częstość występowania hiperprolinemii typu II szacowana jest na 1:700 000 żywych urodzeń, jest to zatem bardzo rzadka choroba 29 .
Napady padaczkowe mogą być skutecznie leczone klasycznymi lekami przeciwpadaczkowymi, ale podaż witaminy B6 lub PLP znacznie poprawia ich kontrolę i zapobiega kolejnym napadom 28 .
Wrodzona hipofosfatazja i hiperfosfatazja
Hipofosfatazja to bardzo rzadka choroba uwarunkowana deficytem białka TNSALP, prowadząca przede wszystkim do zaburzeń w układzie kostnym, występująca już od okresu prenatalnego. Zmiany przypominają deformacje występujące w krzywicy, często prowadzą do znacznego zniekształcenia klatki piersiowej i zaburzeń oddychania z tym związanych. Stężenie fosfatazy alkalicznej jest poniżej wartości referencyjnej dla wieku, a w surowicy stwierdza się podwyższone stężenie PLP. Część pacjentów ma napady padaczkowe oporne na klasyczne leki przeciwpadaczkowe, a ustępujące po podaży witaminy B6 3 .
Białko TNSALP, aby przejść przez barierę krew–mózg, wymaga białek kotwiczących – m.in. PIGO, PIGE. Nieprawidłowe warianty molekularne w genach kodujących te białka również mogą ujawnić się klinicznie jako drgawki odpowiadające na witaminę B6, a biochemicznie charakterystyczne jest podwyższone stężenie fosfatazy alkalicznej w surowicy 3 .
Istnieje również grupa drgawek występujących u noworodków i niemowląt, które ustępują po podaży pirydoksyny, ale nie stwierdza się wśród nich pirydoksynozależności – tzn. po odstawieniu leczenia witaminą B6 nie obserwuje się nawrotu drgawek, rzadziej w tej grupie występuje stan padaczkowy i zazwyczaj rozwój psychoruchowy, a później intelektualny pacjentów jest w granicach normy 30 .
Podsumowanie
Drgawki u noworodków i małych niemowląt mogą mieć bardzo różną etiologię, najczęściej są to zaburzenia wynikające z encefalopatii niedotleniowo-niedokrwiennej, zakażenia uogólnionego, neuroinfekcji czy zaburzeń elektrolitowych. Wrodzone wady metabolizmu są rzadkimi chorobami, szacuje się, że odpowiadają za niespełna 7% encefalopatii padaczkowych ujawniających się w pierwszym roku życia 31 . Część z nich wymaga specyficznego postępowania, a wczesne włączenie odpowiedniej terapii poprawia rokowanie nie tylko co do opanowania napadów, ale również rozwoju pacjentów. Dlatego zawsze przy napadach u noworodków czy małych dzieci, szczególnie o niejasnej etiologii, należy podjąć próbę leczenia witaminą B6 i przeprowadzić diagnostykę w kierunku wrodzonych wad metabolizmu.
Abstract
Pyridoxine-induced seizures
Vitamin B6 has numerous important functions in the human body, e.g. it is a cofactor for a number of enzymes and metabolic pathways. The deficiency of vitamin B6 due to inborn errors of metabolism leads to epileptic encephalopathy which often manifests itself in the neonatal period and early infancy. The resulting seizures are often resistant to conventional antiepileptic drugs, but tend to disappear after a therapeutic dose of vitamin B6 or its active form – pyridoxal phosphate. Whenever a patient is unresponsive to antiepileptic drugs, especially a patient with seizures of unexplained etiology, inborn errors of metabolism should be considered and treatment based on vitamin B6 should be attempted.
- 1. Plecko B. Pyridoxine and pyridoxalphosphate-dependent epilepsies. Handb Clin Neurol 2013;113:1811-7
- 2. Tong Y. Seizures caused by pyridoxine (vitamin B6) deficiency in adults: A case report and literature review. Intractable Rare Dis Res 2014;3(2):52-6
- 3. Wilson MP, Plecko B, Mills PB, Clayton PT. Disorders affecting vitamin B6 metabolism. J Inherit Metab Dis 2019;42(4):629-64
- 4. Mills PB, Struys E, Jakobs C, et al. Mutations in antiquitin in individuals with pyridoxine-dependent seizures. Nat Med 2006;12:307-9
- 5. Coughlin CR, Swanson MA, Spector E, et al. The genotypic spectrum of ALDH7A1 mutations resulting in pyridoxine dependent epilepsy: a common epileptic encephalopathy. J Inherit Metab Dis 2018. https://doi.org/10.1007/s10545-018-0219-7
- 6. Baxter P. Pyridoxine-dependent and pyridoxine-responsive seizures. Dev Med Child Neurol 2001;43:416-20
- 7. Jiao X, Xue J, Gong P, et al. Clinical and genetic features in pyridoxine-dependent epilepsy: a Chinese kohort study. Dev Med Child Neurol 2020;62:315-21
- 8. Basura GJ, Hagland SP, Wiltse AM, Gospe SM. Clinical features and the management of pyridoxine-dependent and pyridoxine-responsive seizures: review of 63 North American cases submitted to a patient registry. Eur J Pediatr 2009;168:697-704
- 9. Coughlin CR, Tseng LA, Abdenur JE, et al. Consensus guidelines for the diagnosis and management of pyridoxine-dependent epilepsy due to α-aminoadipic semialdehyde dehydrogenase deficiency. J Inherit Metab Dis 2021;44(1):178-92
- 10. Mills PB, Footitt EJ, Mills KA, et al. Genotypic and phenotypic spectrum of pyridoxine-dependent epilepsy (ALDH7A1 deficiency). Brain 2010;133:2148-59
- 11. Coughlin CR, Swanson MA, Spector E, et al. The genotypic spectrum of ALDH7A1 mutations resulting in pyridoxine dependent epilepsy: a common epileptic encephalopathy. J Inherit Metab Dis 2019;42:353-61
- 12. Van Karnebeek CD, Tiebout SA, Niermeijer J, et al. Pyridoxinedependent epilepsy: an expanding clinical spectrum. Pediatr Neurol 2016;59:6-12
- 13. Wempe MF, Kumar A, Kumar V, et al. Identification of a novel biomarker for pyridoxine-dependent epilepsy: Implications for newborn screening. J Inherit Metab Dis 2019;42:565-74
- 14. Kuhara T, Akiyama T, Ohse M, et al. Identification of new biomarkers of pyridoxine-dependent epilepsy by GC/MS-based urine metabolomics. Anal Biochem 2020; 604:113739
- 15. Gospe SM, Neonatal vitamin-responsive epileptic encephalopathies. Chang Gung Med J 2010;33:1-12
- 16. Cosnahan AS, Campbell CT. Inborn Errors of Metabolism in Pediatric Epilepsy. J Pediatr Pharmacol Ther 2019;24(5):398-405
- 17. Sharma S, Prasad AN. Inborn errors of metabolism and epilepsy: current understanding, diagnosis and treatment approaches. Int J Mol Sci 2017;18:1384
- 18. Coughlin CR, van Karnebeek CDM, Al-Hertani W, et al. Triple therapy with pyridoxine, arginine supplementation and dietary lysine restriction in pyridoxine-dependent epilepsy: neurodevelopmental outcome. Mol Genet Metab 201;116:35-43
- 19. Mahajnah M, Corderio D, Austin V, et al. A prospective case study of the safety and efficacy of lysine-restricted diet and arginine supplementation therapy in a patient with pyridoxine-dependent epilepsy caused by mutations in ALDH7A1. Pediatr Neurol 2016;60:60-5
- 20. Mercimek-Mahmutoglu S, Cordeiro D, Cruz V, et al. Novel therapy for pyridoxine dependent epilepsy due to ALDH7A1 genetic defect: l-arginine supplementation alternative to lysine-restricted diet. Eur J Paediatr Neurol 2014;18:741-6
- 21. Van Karnebeek CDM, Hartmann H, Jaggumantri S, et al. Lysine restricted diet for pyridoxine-dependent epilepsy: first evidence and future trials. Mol Genet Metab 2012;107:335-44
- 22. Al Teneiji A, Bruun TUJ, Cordeiro D, et al. Phenotype, biochemical features, genotype and treatment outcome of pyridoxinedependent epilepsy. Metab Brain Dis 2017;32:443-51
- 23. Falsaperla R, Vari MS, Toldo I, et al. Pyridoxine-dependent epilepsies: an observational study on clinical, diagnostic, therapeutic and prognostic features in a pediatric cohort. Metab Brain Dis 2018;33:261-9
- 24. Mills PB, Camuzeaux SS, Footitt EJ, et al. Epilepsy due to PNPO mutations: genotype, environment and treatment affect presentation and outcome. Brain 2014;137(5):1350-60
- 25. Hatch J, Coman D, Clayton P, et al. Normal neurodevelopmental outcomes in PNPO deficiency: a case series and literature review. JIMD Rep 2016;26:91-7
- 26. Darin N, Reid E, Prunetti L, et al. Mutations in PROSC disrupt cellular pyridoxal phosphate homeostasis and cause vitamin-B6-dependent epilepsy. Am J Hum Genet 2016;99: 1325-37
- 27. Johnstone DL, Al-Shekaili HH, Tarailo-Graovac M, et al. PLPHP deficiency: clinical, genetic, biochemical, and mechanistic insights. Brain 2019;142(3):542-59
- 28. Mitsubuchi H, Nakamura K, Matsumoto S, Endo F. Biochemical and clinical features of hereditary hyperprolinemia. Pediatr Int 2014;56(4):492-6
- 29. Motte J, Fisse AL, Grüter T, et al. Novel variants in a patient with late-onset hyperprolinemia type II: diagnostic key for status epilepticus and lactic acidosis. BMC Neurol 2019;19(1):345
- 30. Riikonen R, Mankinen K, Gaily E. Long-term outcome in pyridoxine-responsive infantile epilepsy. Eur J Paediatr Neurol 2015;19:647-51
- 31. Mercimek-Mahmutoglu S, Patel J, Cordeiro D, et al. Diagnostic yield of genetic testing in epileptic encephalopathy in childhood. Epilepsia 2015;56(5):707-16
Następny artykuł:
Diagnostyka różnicowa i rokowanie w ilościowych zaburzeniach świadomości


 Dodaj do ulubionych
Dodaj do ulubionych
