



Co znajdziesz w artykule?
- Charakterystyka obrazu klinicznego i przebiegu chorób autozapalnych w populacji pediatrycznej
- Diagnostyka wrodzonych zespołów gorączek nawrotowych – kryteria Eurofever/PRINTO z 2019 roku
- Zastosowanie leków biologicznych w leczeniu chorób autozapalnych
Spis treści
Choroby autozapalne stanowią stosunkowo nową grupę chorób, funkcjonującą oficjalnie w medycynie od 1997 roku, kiedy to został zidentyfikowany gen odpowiedzialny za występowanie rodzinnej gorączki śródziemnomorskiej (FMF – familial Mediterranean fever). Pod koniec lat 90. XX wieku opisano dwie inne autozapalne jednostki chorobowe: zespół hiperimmunoglobulinemii D (HIDS – hyperimmunoglobulin D syndrome) oraz zespół gorączki nawrotowej zależny od receptora czynnika martwicy nowotworów (TRAPS –
tumor necrosis factor [TNF] receptor-associated periodic syndrome). W ciągu ostatnich 20 lat opisano kolejne zespoły chorobowe o podłożu autozapalnym charakteryzujące się dużą heterogennością i na ogół wczesnym początkiem objawów 1 . Patofizjologię procesu autozapalnego cechują przewlekły przebieg oraz uszkodzenia o długoterminowym charakterze. Od kilku lat dostępne są leki skutecznie kontrolujące mechanizmy autozapalenia oraz główne objawy, co umożliwia pacjentom dotkniętym tymi genetycznie uwarunkowanymi chorobami zachowanie akceptowalnej jakości życia, chociaż wciąż nie ma skutecznej terapii większości ciężkich manifestacji. Na ogół leczenie farmakologiczne jest oparte na grupie leków biologicznych blokujących cząsteczki zaangażowane w proces zapalny, które są produkowane w nasilonej ilości w ostrej fazie epifenomenu zapalnego 2 .
Definicja, obraz kliniczny i patofizjologia chorób autozapalnych
Choroby autozapalne stanowią grupę chorób, które ujawniają się w wyniku dysfunkcji lub dysregulacji układu odporności wrodzonej i mogą powodować poważne powikłania oraz ryzyko zgonu poprzez zajęcie wielu narządów. Ta nowa grupa chorób różni się od standardowej koncepcji chorób z autoimmunizacji, które są relatywnie dobrze poznane w zakresie podstawowych mechanizmów, defektem odporności wrodzonej i brakiem krążących autoprzeciwciał w krwiobiegu. Termin „choroby autozapalne” został po raz pierwszy użyty przez amerykańskie National Institutes of Health na określenie systemowego zapalenia charakteryzującego się nieprowokowanymi atakami, bez obecności przeciwciał i antygenowo swoistych limfocytów T. Z kolei Kastneri i wsp. w 2010 roku zdefiniowali choroby autozapalne jako choroby cechujące się nadmiernie nasilonym zapaleniem mediowanym głównie przez komórki i cząsteczki układu odporności wrodzonej u osób predysponowanych. W 2017 roku Wekell i wsp. zmodyfikowali tę definicję, określając choroby autozapalne jako: „choroby immunologicznie uwarunkowane z nadmiernym zapaleniem spowodowanym przez dysregulację komórek i cząsteczek wrodzonej odporności, przy znacznej predyspozycji gospodarza, ale często również z aktywacją układu odporności nabytej i dysfunkcją immunologiczną (podatność na infekcje), autoimmunizacją lub niekontrolowanym nadmiernym zapaleniem”. Ostateczna definicja chorób autozapalnych została sformułowana przez Paediatric Rheumatology INternational Trials Organisation (PRINTO) w 2018 roku, zgodnie z którą to „jednostki kliniczne spowodowane defektem bądź dysregulacją wrodzonej odpowiedzi immunologicznej, charakteryzujące się nawracającym lub ciągłym zapaleniem (podwyższone wskaźniki ostrej fazy) i brakiem pierwotnej roli patogennej układu odporności nabytej (brak autoreaktywnych limfocytów T lub produkcji autoprzeciwciał)”.
Choroby autozapalne mają różny obraz kliniczny w zależności od specyficznej mutacji genetycznej odpowiedzialnej za chorobę i cechują się występowaniem zapalenia powstałego w wyniku uwalniania specyficznych cytokin prozapalnych (głównie interleukiny [IL] 1, TNFα czy interferonów [IFN] α, β). Początek objawów chorobowych obserwuje się już w pierwszych miesiącach/latach życia z gorączką, zajęciem stawów, skóry oraz obecnością markerów serologicznych. Objawy choroby autozapalnej pojawiają się nagle, w stanie zdrowia, z charakterystyczną wysoką ciepłotą ciała (do 39-40°C) 3, 4 .
Klasyfikacja chorób autozapalnych ze względu na patogenezę
Podział chorób autozapalnych w zależności od czynnika patofizjologicznego przedstawiono w tabeli 1.
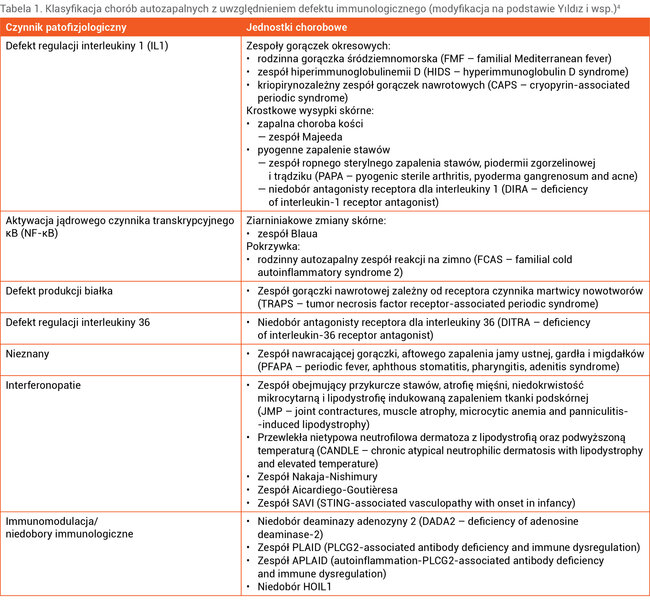
Tabela 1. Klasyfikacja chorób autozapalnych z uwzględnieniem defektu immunologicznego (modyfikacja na podstawie Yıldız i wsp.)4
Zespoły gorączek nawrotowych
Zespoły gorączek nawrotowych (periodic fever syndromes) to heterogenna grupa układowych chorób autozapalnych, które charakteryzują się nawracającymi epizodami zapalenia bez związku z infekcją, procesem autoimmunizacyjnym czy z chorobą nowotworową. U podłoża większości chorób z tej grupy leżą mutacje genów kodujących białka związane ze szlakami reakcji zapalnej, apoptozą i wytwarzaniem cytokin, prowadzące do zaburzenia mechanizmów odporności wrodzonej. Większość chorób z tej grupy ma początek w wieku rozwojowym. Dominującym objawem są epizody gorączki, której towarzyszą zmiany skórne, zapalenie stawów, dolegliwości ze strony przewodu pokarmowego (ból brzucha, biegunka). W czasie zaostrzenia choroby stwierdza się istotnie zwiększone wartości wskaźników stanu zapalnego oraz stężenia cytokin prozapalnych. W okresie między rzutami objawy ustępują, a wyniki badań laboratoryjnych zazwyczaj się normalizują.
Zespoły gorączek nawrotowych są wyzwaniem diagnostycznym dla klinicysty. Kluczowe jest przeprowadzenie szerokiej diagnostyki różnicowej uwzględniającej infekcje, choroby z autoimmunizacji czy rozrostowe. Przed ustaleniem rozpoznania wskazane są obserwacja pacjenta przez minimum 6 miesięcy, notowanie czasu trwania gorączki i okresów bezobjawowych, udokumentowanie wzrostu wartości wskaźników stanu zapalnego w momencie rzutu choroby. Istotne jest dokładne badanie podmiotowe pacjenta, uwzględniające wiek ujawnienia się objawów, pochodzenie etniczne chorego oraz występowanie podobnych objawów w rodzinie.
Do najczęściej rozpoznawanych zespołów gorączek nawrotowych należą:
- rodzinna gorączka śródziemnomorska (FMF)
- zespół gorączki nawrotowej zależny od receptora dla TNF (TRAPS)
- zespół hiperimmunoglobulinemii D (HIDS)
- kriopirynozależny zespół gorączek nawrotowych (CAPS)
- zespół nawracającej gorączki, aftowego zapalenia jamy ustnej, gardła i migdałków (PFAPA).
Rodzinna gorączka śródziemnomorska
Jest najczęstszą monogenową chorobą autozapalną na świecie. Największą liczbę zachorowań odnotowuje się w basenie Morza Śródziemnego, tj. wśród Arabów, Turków, Ormian, Żydów sefardyjskich. W innych populacjach FMF należy do rzadkości. U większości chorych pierwsze objawy pojawiają się przed 10 rokiem życia. Choroba jest dziedziczona autosomalnie recesywnie. U podłoża FMF leżą mutacje genu MEFV kodującego pirynę. Zmieniona cząsteczka tego białka staje się nieefektywnym inhibitorem odpowiedzi zapalnej, co prowadzi do nasilenia apoptozy i produkcji interleukiny 1β (IL1β). Rzut choroby trwa zwykle 1-4 dni, często jest poprzedzony infekcją, urazem lub stresem. Wysokiej gorączce towarzyszy zapalenie błon surowiczych, najczęściej otrzewnej, mogące imitować objawy tzw. ostrego brzucha. Rzadziej występują: zapalenie opłucnej (objawiające się zazwyczaj bólem w klatce piersiowej), osierdzia, moszny (objawy sugerujące skręt jądra), zapalenie opon mózgowo-rdzeniowych. U blisko 50% chorych stwierdza się przemijające zapalenie dużych stawów (najczęściej kolanowego, biodrowego, skokowego). U 25% pacjentów na grzbietowej powierzchni stóp i stawu skokowego pojawia się przemijająca, różopodobna wysypka. W początkowym okresie FMF możliwe jest występowanie gorączki jako izolowanego objawu choroby.
Rozpoznanie FMF ustala się na podstawie nowych kryteriów klinicznych Eurofever/PRINTO, które obejmują stwierdzenie mutacji w genie MEFV oraz spełnienie co najmniej jednego z 4 następujących kryteriów: rzuty choroby trwające 1-3 dni, zapalenie stawów, ból w klatce piersiowej, ból brzucha. W przypadku braku mutacji patognomonicznych w genie MEFV wskazane jest spełnienie minimum 2 z powyższych kryteriów.
Badania dodatkowe pełnią funkcję pomocniczą w diagnostyce FMF. W okresie rzutu choroby zazwyczaj stwierdza się: podwyższone wartości wskaźników stanu zapalnego, nieznaczną leukocytozę, nadpłytkowość, zwiększone stężenia immunoglobulin. Istotne jest regularne wykonywanie badania ogólnego moczu, gdyż białkomocz występujący w okresie między rzutami może sugerować rozwój powikłania w postaci amyloidozy (!).
FMF wymaga różnicowania z innymi zespołami gorączek nawrotowych, chorobami autoimmunizacyjnymi czy rozrostowymi.
Podstawowym lekiem stosowanym w leczeniu FMF jest kolchicyna. Jest ona skuteczna w zapobieganiu rzutom choroby oraz powstawaniu amyloidozy. Leczeniem drugiej linii są inhibitory IL1: anakinra i kanakinumab. Chorzy z FMF powinni być objęci opieką interdyscyplinarnego zespołu specjalistów posiadających doświadczenie w leczeniu tej jednostki chorobowej, składającego się z reumatologa koordynującego leczenie oraz dodatkowo w zależności od objawów pacjenta z: gastrologa, nefrologa, ortopedy i fizjoterapeuty.
Poważnym powikłaniem FMF jest amyloidoza, najczęściej nerek, która może prowadzić do ich niewydolności. U pacjentów z FMF mogą również występować zaburzenia płodności wtórne do amyloidozy jąder lub nawracające zapalenia otrzewnej 1, 4, 5, 6 .
Zespół gorączki nawrotowej zależny od receptora dla TNF
TRAPS jest zespołem uwarunkowanym genetycznie spowodowanym mutacją w obrębie białka kodującego receptor 1A TNF (TNFRSF1A). Choroba dziedziczona jest autosomalnie dominująco. Po raz pierwszy została opisana w 1982 roku u 16 członków rodziny irlandzko-szkockiego pochodzenia. Początkowo sądzono, że jest to wariant FMF, jednak nie obserwowano satysfakcjonującej odpowiedzi na leczenie kolchicyną. Obecnie przypadki tej jednostki chorobowej są stwierdzane we wszystkich grupach etnicznych na całym świecie.
Objawy kliniczne TRAPS są zróżnicowane u poszczególnych pacjentów. Pierwsze symptomy zazwyczaj pojawiają się we wczesnym dzieciństwie, jednak w łagodniejszych wariantach choroby mogą wystąpić później, nawet po 30 roku życia. Epizody gorączki nawracają w nieregularnych odstępach, u niektórych chorych co 3-4 tygodnie, u innych pojawiają się tylko 2 razy do roku. Wysokiej gorączce towarzyszą: ból brzucha, dolegliwości bólowe/zapalenie stawów i mięśni, rumieniowata wysypka, zapalenie spojówek, obrzęk okolicy oczodołów. Zapalenie błon surowiczych może manifestować się jako nawracające epizody zapalenia osierdzia, otrzewnej i opłucnej.
Rozpoznanie TRAPS ustala się na podstawie nowych kryteriów klinicznych Eurofever/PRINTO, które obejmują stwierdzenie mutacji w genie TNFRSF1A oraz spełnienie co najmniej jednego z 5 następujących kryteriów: czas trwania rzutów choroby ≥7 dni, ból mięśni, wędrująca wysypka, obrzęk okolicy oczodołów, obciążony wywiad rodzinny. W przypadku braku potwierdzenia mutacji w obrębie genu TNFRSF1A wskazane jest spełnienie minimum 2 z powyższych kryteriów.
Podczas rzutu choroby obserwuje się istotny wzrost wskaźników ostrej fazy (odczynu Biernackiego [OB], stężenia białka C-reaktywnego [CRP – C-reactive protein], amyloidu A), w morfologii krwi obwodowej leukocytozę z przewagą neutrofilów, nadpłytkowość oraz niedokrwistość normocytarną. Między rzutami wskaźniki stanu zapalnego mogą być nieznacznie podwyższone. Wskazane jest regularne wykonywanie badań oceniających funkcję nerek (stężenie kreatyniny, badanie ogólne moczu – białkomocz [!]).
Choroba wymaga różnicowania z: infekcjami, chorobami układowymi tkanki łącznej, postacią układową młodzieńczego idiopatycznego zapalenia stawów, chorobami rozrostowymi oraz innymi zespołami gorączek nawrotowych.
Zgodnie z wytycznymi grupy Single Hub and Access point for paediatric Rheumatology in Europe (SHARE) w leczeniu TRAPS można stosować:
- niesteroidowe leki przeciwzapalne (NLPZ) – objawowo podczas rzutu gorączki
- glikokortykosteroidy (GKS) systemowe – w celu przerwania rzutu choroby
- inhibitory IL1 (anakinrę, kanakinumab) oraz TNF (etanercept) – u pacjentów z częstymi epizodami gorączki oraz zapaleniem utrzymującym się mimo stosowania GKS systemowych.
Pacjent z TRAPS powinien być leczony przez interdyscyplinarny zespół pod kierownictwem reumatologa bądź immunologa dziecięcego posiadających doświadczenie w leczeniu chorób autozapalnych.
Najcięższym następstwem nieleczonego TRAPS jest amyloidoza. U 80% chorych z tym powikłaniem dochodzi do niewydolności nerek; w innych narządach złogi amyloidu występują rzadko 4, 5, 7 .
Deficyt kinazy mewalonianowej/ zespół hiperimmunoglobulinemii D
To rzadka jednostka chorobowa, która wprawdzie może występować we wszystkich grupach etnicznych, jednak najczęściej opisywana jest u mieszkańców północnej i zachodniej Europy. Mediana wieku rozpoznania HIDS wynosi 6 miesięcy. Występuje z jednakową częstością u obu płci. Choroba dziedziczona autosomalnie recesywnie, jest spowodowana mutacją genu kinazy mewalonianowej (MVK – mevalonate kinase), którego produkt jest kluczowym enzymem biosyntezy cholesterolu i izoprenoidów. Deficyt MVK prowadzi do aktywacji inflamasomu pirynowego, dochodzi również do zwiększonej syntezy IL1 poprzez aktywację kaspazy 1.
Gorączka w przebiegu HIDS trwa zazwyczaj 3-6 dni, nawraca co 4-8 tygodni i pojawia się już w wieku niemowlęcym. Inne typowe objawy to: bolesne powiększenie węzłów chłonnych szyjnych, objawy ze strony przewodu pokarmowego (ból brzucha, wymioty, biegunka), zajęcie skóry i błon śluzowych (zapalenie jamy ustnej, zapalenie gardła, bolesna wysypka drobno- i gruboplamista), ból/zapalenie stawów obwodowych. W przypadkach gdy aktywność MVK jest wyjątkowo mała (<10%), rozwijają się dodatkowo powikłania kwasicy mewalonianowej, takie jak: mikrocefalia, opóźnienie rozwoju i dysmorfia twarzy.
Rozpoznanie HIDS ustala się na podstawie nowych kryteriów klinicznych Eurofever/PRINTO, które obejmują konieczność stwierdzenia mutacji w obrębie genu MVK oraz spełnienia minimum jednego z 3 następujących kryteriów: objawy ze strony przewodu pokarmowego, limfadenopatia szyjna, aftowe zapalenie jamy ustnej.
Podczas rzutu choroby obserwuje się znaczne zwiększenie wartości wskaźników stanu zapalnego, leukocytozę, podwyższone stężenie amyloidu A. U około 80% pacjentów stwierdza się wzrost stężenia IgD (>100 j.m./ml), z towarzyszącym zazwyczaj zwiększeniem stężenia IgA. U 93% pacjentów podczas rzutu choroby obserwuje się zwiększone wydalanie kwasu mewalonianowego z moczem. Pomocne w diagnostyce jest badanie genetyczne w kierunku mutacji genu MVK.
HIDS wymaga różnicowania z: infekcjami, chorobami autoimmunologicznymi, rozrostowymi i innymi zespołami gorączek nawrotowych.
W leczeniu objawowym podczas rzutu choroby można stosować NLPZ oraz GKS systemowe. Jeśli nie uzyskuje się zadowalającej odpowiedzi klinicznej na GKS, wskazane jest włączenie do leczenia inhibitora IL1 (anakinry, kanakinumabu) lub TNF (etanerceptu). Kolchicyna nie wykazuje skuteczności w leczeniu HIDS. Chory na HIDS powinien być objęty opieką interdyscyplinarną, koordynowaną przez immunologa specjalizującego się w leczeniu zespołów gorączek nawrotowych.
Epizody gorączki pojawiają się przez całe życie, choć ich częstość jest największa w dzieciństwie. Możliwy jest rozwój amyloidozy 3, 4, 7 .
Kriopirynozależne zespoły gorączek nawrotowych
Wyróżnia się 3 fenotypy kliniczne CAPS:
- rodzinny autozapalny zespół reakcji na zimno (FCAS)
- zespół Muckle’a-Wellsa (MWS – Muckle-Wells syndrome)
- przewlekły niemowlęcy zespół neurologiczno-skórno-stawowy (CINCA – chronic infantile neurologic cutaneous and articular syndrome), zwany także noworodkową chorobą wieloukładową (NOMID – neonatal-onset multisystem inflammatory disease).
Wszystkie powyższe zespoły należą do chorób uwarunkowanych genetycznie, które charakteryzują się wczesnym początkiem epizodów gorączki z towarzyszącymi cechami uogólnionego zapalenia, pokrzywką, różnie wyrażonymi objawami ze strony narządu ruchu oraz zapaleniem spojówek. Zespół FCAS jest najłagodniejszą postacią choroby, natomiast NOMID/CINCA najcięższą.
CAPS to rzadka choroba, rozpoznawana w Europie u 1-2 pacjentów na 1 mln mieszkańców. FCAS i MWS mogą występować rodzinnie, fenotyp NOMID/CINCA jest spowodowany mutacją de novo. Przyczyną choroby jest mutacja genu NLRP3 kodującego kriopirynę. Zmieniona sekwencja tego białka prowadzi do nadmiernej aktywacji inflamasomu NLRP3, która skutkuje niekontrolowaną produkcją IL1β. Choroba jest dziedziczona autosomalnie dominująco.
Objawy kliniczne zależą od postaci:
- FCAS – objawy pojawiają się od 30 min do 6 godz. po ekspozycji na zimno i ustępują w ciągu 48 godz. Typowe spektrum kliniczne choroby obejmuje: gorączkę, pokrzywkę, zapalenie spojówek, ból bądź obrzęk stawów. Początek objawów obserwuje się już w wieku niemowlęcym
- MWS – cięższa forma choroby; przebiega z gorączką, uogólnioną pokrzywką, zapaleniem spojówek, bólem i zapaleniem stawów, którym towarzyszy pogłębiający się niedosłuch odbiorczy
- NOMID/CINCA – jedna z najcięższych monogenowych chorób autozapalnych. Wśród objawów dominują: utrzymująca się gorączka, pokrzywka, zapalenie błony naczyniowej, głuchota odbiorcza, oraz dochodzi do rozwoju destrukcyjnego zapalenia stawów z przerostem chrząstek stawowych i następczym kostnieniem (głównie rzepek). Dodatkowo występuje szeroki zakres objawów ze strony ośrodkowego układu nerwowego: przewlekłe, aseptyczne zapalenie opon mózgowo-rdzeniowych, atrofia nerwu wzrokowego, wodogłowie, opóźnienie umysłowe.
CAPS rozpoznaje się na podstawie nowych kryteriów Eurofever/PRINTO, które obejmują obecność mutacji w genie NLRP3 i jednego z wymienionych objawów, takich jak: pokrzywka, objawy zapalne ze strony narządu wzroku (zapalenie spojówek, błony naczyniowej, nadtwardówki), neurosensoryczna utrata słuchu. W przypadku braku potwierdzonej mutacji w obrębie genu NLRP3 konieczne jest spełnienie minimum 2 z wyżej wymienionych kryteriów.
Rozpoznanie kliniczne powinno być potwierdzone diagnostyką molekularną w kierunku mutacji genu NLRP3. U części pacjentów z CAPS stwierdza się mozaicyzm w obrębie tego genu, który może nie zostać wykryty w standardowym badaniu genetycznym. W testach laboratoryjnych obserwuje się zwiększone wartości wskaźników stanu zapalnego (OB, CRP); wskazane jest również monitorowanie stężenia amyloidu A, który jest wczesnym markerem rozwoju amyloidozy. U pacjentów z zespołem NOMID/CINCA w badaniu metodą jądrowego rezonansu magnetycznego można zobrazować poszerzenie komór mózgu oraz zwiększoną przestrzeń podtwardówkową. W badaniu płynu mózgowo-rdzeniowego stwierdza się pleocytozę z podwyższonym odsetkiem granulocytów oraz zwiększonym stężeniem białka. W MWS oraz NOMID/CINCA wskazane jest wykonywanie audiometrii. W przypadku wątpliwych diagnostycznie zmian skórnych przydatna może być jej biopsja.
CAPS należy różnicować z: innymi dziedzicznymi gorączkami nawrotowymi, zakażeniami wrodzonymi oraz młodzieńczym idiopatycznym zapaleniem stawów o początku uogólnionym.
Podstawą leczenia CAPS jest stosowanie inhibitorów IL1 (anakinry, kanakinumabu). W leczeniu objawowym wskazane jest przyjmowanie NLPZ oraz GKS systemowych. Nie ma dowodów na działanie leków modyfikujących przebieg choroby w tym wskazaniu. Opieka nad pacjentem z CAPS powinna być wielodyscyplinarna, a dokładny skład zespołu specjalistów zależy od fenotypu klinicznego choroby oraz objawów występujących u pacjenta.
Rokowanie zależy od postaci choroby. U pacjentów z FCAS jest ono dobre, ich jakość życia nie odbiega znacząco od jakości życia zdrowej populacji, jeśli unikają ekspozycji na zimno. U 25% chorych na MWS rozwija się amyloidoza, postępuje również niedosłuch odbiorczy. W zespołach NOMID/CINCA rokowanie jest najgorsze, u nieleczonych pacjentów śmiertelność jest wysoka. Jakość życia zależy od ciężkości uszkodzenia stawów, uszkodzenia narządu wzroku i słuchu. Przewlekłe zapalenie opon mózgowo-rdzeniowych decyduje o odległym rokowaniu 4, 5, 8, 9 .
Zespół nawracającej gorączki, aftowego zapalenia jamy ustnej, gardła i migdałków
Zespół PFAPA został opisany po raz pierwszy w 1987 roku. Jest najczęstszą idiopatyczną chorobą autozapalną wieku rozwojowego, a zachorowalność w populacji europejskiej jest szacowana na 2,3/10 000. Częściej dotyczy chłopców (68%) niż dziewcząt. Objawy pojawiają się na ogół między 2 a 5 rokiem życia i ulegają remisji w drugiej dekadzie życia. Rozpoznawalność zespołu dynamicznie wzrasta. Dotąd nie ustalono jednoznacznie podłoża genetycznego PFAPA, choć występowanie objawów zespołu u członków rodzin pacjentów może świadczyć o wielogenowym dziedziczeniu choroby. Wiedza na temat etiopatogenezy PFAPA jest ograniczona, jednak tak jak w przebiegu innych zespołów autozapalnych obserwuje się nadmierną aktywację inflamasomów i zwiększoną produkcję IL1β.
Objawy kliniczne PFAPA:
- gorączka trwająca zazwyczaj 4-6 dni i nawracająca co 3-5 tygodni; pojawia się nagle, z reguły przekracza 39°C, słabo reaguje na leki przeciwgorączkowe (periodic fever)
- aftowe zapalenie jamy ustnej (aphthous stomatitis)
- zapalenie gardła, powiększone migdałki podniebienne z cechami zapalenia, ich wygląd może sugerować infekcję wirusową (mononukleozę) bądź bakteryjną (anginę paciorkowcową) (pharyngitis)
- limfadenopatia szyjna (cervical adenitis).
Rozpoznanie PFAPA ustala się na podstawie kryteriów Marshalla i Edwardsa 9 lub Eurofever/PRINTO 4 , które przedstawiono w tabeli 2.

Tabela 2. Kryteria rozpoznania zespołu nawracającej gorączki, aftowego zapalenia jamy ustnej, gardła i migdałków (PFAPA)4,9
Podczas rzutu choroby w morfologii obserwuje się leukocytozę z odmłodzeniem obrazu białokrwinkowego oraz podwyższone wartości OB i CRP przy prawidłowym lub nieznacznie podwyższonym stężeniu prokalcytoniny. W okresie między rzutami wyniki badań laboratoryjnych ulegają normalizacji. Kluczowa dla rozpoznania jest analiza dotychczasowego przebiegu choroby, długości i częstotliwości gorączek, pomocne są w tym prowadzone przez rodziców dzienniczki obserwacji objawów. W przypadkach wątpliwych wskazane jest poszerzenie diagnostyki o badanie genetyczne w kierunku innych zespołów gorączek nawrotowych.
Zespół PFAPA wymaga różnicowania przede wszystkim z infekcjami – mononukleozą zakaźną czy anginą paciorkowcową. Istotne jest kilkukrotne wykonanie morfologii krwi obwodowej w celu wykluczenia cyklicznej neutropenii. Należy również wykluczyć zakażenie układu moczowego, wykonując badanie ogólne oraz posiew moczu co najmniej przy dwóch nawrotach choroby. Wskazane są konsultacja stomatologiczna i badanie echokardiograficzne (wykluczenie ognisk zapalnych).
Leczeniem pierwszego rzutu jest prednizon w dawce 1 mg/kg podany na początku rzutu choroby. Terapia hamuje objawy rzutu u ponad 80% pacjentów, nie zapobiega jednak kolejnym nawrotom, a u niektórych dzieci wpływa na skrócenie okresów bezobjawowych między rzutami. Skuteczna jest również tonsillektomia, dzięki której u większości pacjentów obserwuje się pełną remisję choroby. Pacjenci z zespołem PFAPA powinni pozostawać pod opieką immunologa zajmującego się leczeniem tej jednostki.
Rokowanie u pacjentów z zespołem PFAPA jest dobre, choroba ulega samoistnej remisji w drugiej dekadzie życia. W przebiegu PFAPA nie dochodzi do rozwoju amyloidozy 4, 5, 8, 9, 10, 11 .
Podsumowanie
- Choroby autozapalne są coraz częściej rozpoznawanymi jednostkami chorobowymi w codziennej praktyce klinicznej, spowodowanymi głównie zaburzeniami układu odporności wrodzonej z udziałem IL1β, IFN i NF-κB.
- Diagnostyka wrodzonych zespołów gorączek nawrotowych powinna opierać się na kryteriach Eurofever/PRINTO z 2019 roku.
- Podstawą leczenia większości jednostek chorobowych z tej grupy są GKS systemowe, ponadto coraz częściej w terapii stosuje się leki biologiczne – głównie inhibitory IL1 i TNF.
- Długoterminowym powikłaniem chorób autozapalnych może być amyloidoza (wyjątek stanowi zespół PFAPA, w którym rokowanie jest dobre).
- Pacjenci z chorobami autozapalnymi wymagają opieki interdyscyplinarnej, w zależności od prezentowanych objawów.
Abstract
Autoinflammatory diseases in children
There is a well-known link between innate and adaptive immune responses, which constitute the distinctive origins of several diseases, many of which may be the consequence of the loss of balance between these two responses. Autoinflammation, as an autoimmune response, is due to the excessive activation of the immune system and shows a clinical phenotype characterized by alternating periods of exacerbation and remission. Hereditary autoinflammatory diseases are an expanding set of disorders characterized by dysregulation of the innate immune system, largely mediated by interleukin-1β, interferon α, β and nuclear factor–kB. For physicians, the challenge lies in diagnosing these rare conditions. Many of the syndromes involve symptoms that mimic allergic and immunodeficiency disorders. The constellation of fevers, rashes, and mucosal symptoms in many of the disorders suggests that the immunologist is the appropriate specialist for managing these patients. Early diagnosis and initiation of therapy is critical to reducing morbidity and mortality in these rare patients. This review focuses on the expanding spectrum of autoinflammatory diseases, diagnosis, and understanding the available options of treatment.
- 1. Livneh A, Drenth JP, Klasen IS, et al. Familial Mediterranean fever and hyperimmunoglobulinemia D syndrome: two diseases with distinct clinical, serologic and genetic features. J Rheumatol 1997;24(8):1558-63
- 2. Kallinich T. Regulating against the dysregulation: new treatment options in autoinflammation. Semin Immunopathol 2015;37(4):429-37
- 3. Marcuzzi A, Melloni E, Zauli G, et al. Autoinflammatory diseases and cytokine storms-imbalances of innate and adaptative immunity. Int J Mol Sci 2021;22(20):11241
- 4. Yıldız M, Haşlak F, Adrovic A, et al. Autoinflammatory diseases in childhood. Balkan Med J 2020;37(5):236-46
- 5. Gattorno M, Hofer M, Federici S, et al.; Eurofever Registry and the Paediatric Rheumatology International Trials Organisation (PRINTO). Classification criteria for autoinflammatory recurrent fevers. Ann Rheum Dis 2019;78(8):1025-32
- 6. Hentgen V, Grateau G, Kone-Paut I, et al. Evidence-based recommendations for the practical management of familial Mediterranean fever. Semin Arthritis Rheum 2013;43(3):387-91
- 7. Cudrici C, Deuitch N, Aksentijevich I. Revisiting TNF receptor-associated periodic syndrome (TRAPS): Current perspectives. Int J Mol Sci 2020;21(9):3263
- 8. Sag E, Bilginer Y, Ozen S. Autoinflammatory diseases with periodic fevers. Curr Rheumatol Rep 2017;19(7):41
- 9. Ter Haar NM, Oswald M, Jeyaratnam J, et al. Recommendations for the management of autoinflammatory diseases. Ann Rheum Dis 2015;74(9):1636-44
- 10. Thomas KT, Feder HM Jr, Lawton AR, et al. Periodic fever syndrome in children. J Pediatr 1999;135(1):15-21
- 11. Wolska-Kuśnierz B, Mikołuć B, Motkowski R i wsp. Zespół PFAPA – wspólne wyzwanie dla pediatry, immunologa i laryngologa. Stand Med Pediatr 2013;10:794-800
Pierwszy artykuł:

 Dodaj do ulubionych
Dodaj do ulubionych
