



Co znajdziesz w artykule?
Atopowe zapalenie skóry (AZS) jest najczęstszą przewlekłą chorobą zapalną skóry. U chorych na AZS objawy kliniczne często wyprzedzają rozwój astmy i alergii na pokarmy. Ostatnie badania koncentrujące się na etiopatogenezie AZS wykazały zaburzenia w procesie końcowego różnicowania komórek naskórka, co stanowi podstawę do rozwoju dysfunkcji strukturalnej oraz funkcjonalnej warstwy rogowej naskórka. Prowadzi to z kolei do ułatwionej penetracji alergenów środowiskowych w głąb naskórka i rozwoju układowego uczulenia IgE-zależnego.
Skóra chorego na AZS wykazuje też wyraźną predyspozycję do kolonizacji lub nadkażeń bakteryjnych, ze szczególnym udziałem patogennego mikroba, jakim jest Staphylococcus aureus oraz herpes simplex virus.
Przyczyny zaburzeń w zakresie bariery skórnej są bardzo złożone i obejmują zarówno czynniki genetyczne, środowiskowe, jak i czynniki immunologiczne. Właśnie ta złożoność rozmaitych czynników warunkujących zaburzenia bariery skórnej odpowiada za kliniczną heterogenność AZS w zakresie czasu pojawienia się pierwszych objawów, ciężkości przebiegu i naturalnej historii choroby.
Niedawno opublikowane wyniki badań wykazały, że wczesna profilaktyka emolientowa może stanowić ochronę bariery skórnej. Już pierwsze, aktywne zmiany skórne wymagają zarówno leczenia miejscowego, jak i aktywacji układu odpornościowego na drodze ogólnej. Dobór odpowiedniej strategii terapeutycznej jest w tym schorzeniu kluczowy. Wczesna interwencja może poprawiać długoterminowe wyniki stosowanego leczenia AZS i ograniczać rozwój ogólnego uczulenia IgE-zależnego, które może być następnie związane z pojawieniem się alergicznych chorób w zakresie układu pokarmowego oraz oddechowego.
Spis treści
Atopowe zapalenie skóry (AZS) jest najczęściej rozpoznawaną, przewlekłą, zapalną chorobą skóry. 1, 2 Ostatnio wykazano silny związek pomiędzy występowaniem różnorodnych schorzeń o podłożu zaburzeń psychicznych a rozwojem objawów klinicznych AZS, co wyraźnie wskazuje na konieczność uwzględnienia szeroko pojętej psychoterapii w planowaniu indywidualnie dobranej konstrukcji działań terapeutycznych zarówno w odniesieniu do samego pacjenta, jak i jego rodziny. 3 U chorych na AZS obserwuje się
też rozwój uczulenia w zakresie pokarmów oraz klinicznych objawów astmy. 4, 5 Wydaje się, że nieprawidłowa struktura i funkcja bariery naskórkowej u chorych na AZS warunkuje ułatwioną penetrację alergenów środowiskowych przez warstwę rogową naskórka i w ten sposób dochodzić może do rozwoju uczulenia IgE-zależnego w odniesieniu do alergenów pokarmowych i powietrznopochodnych (alergia na pokarmy, astma oskrzelowa). Ponieważ obecnie nie dysponujemy możliwościami wyleczenia pacjenta z alergii pokarmowej czy astmy, jak najdokładniejsze wyjaśnienie mechanizmów leżących u podstaw rozwoju AZS wydaje się niezwykle istotnym elementem w zakresie tzw. wczesnej interwencji.
AZS jest złożoną chorobą o silnie wyrażonym podłożu genetycznym wpływającym na charakterystykę pierwotnej i nabytej odpowiedzi immunologicznej. Poza tym niezwykle istotną rolę odgrywają różnorodne czynniki środowiskowe, takie jak:
- ekspozycja na alergeny,
- czynniki drażniące,
- drobnoustroje,
- dieta,
- stres,
- zanieczyszczenia powietrza. 6, 7, 8, 9, 10
Dotychczas AZS postrzegane było jako dermatoza o relatywnie jednorodnej charakterystyce, ale w świetle ostatnich badań należy stwierdzić, że istnieje konieczność rozróżnienia odmiennych fenotypów i endotypów AZS (tabela 1) 11, 12 w podobny sposób, w jaki dokonano tego w przypadku astmy oraz rhinosinusitis. Wyodrębniono bowiem podgrupy w oparciu o takie elementy jak:
- charakterystyka rozwoju pierwszych objawów klinicznych,
- biomarkery,
- polaryzacja immunologiczna,
- warianty genowe,
- naturalny przebieg choroby. 13, 14, 15

Tabela 1. Podstawowe fenotypy atopowego zapalenia skóry.
Wydaje się, że szczególnie istotne jest określenie poszczególnych ścieżek polaryzacji immunologicznej ze względu na to, że znamy leki biologiczne o działaniu ściśle ukierunkowanym na poszczególne ścieżki immunologiczne, takie jak ścieżka Th2 i Th22, oraz leki działające na wybrane cytokiny i mediatory biorące udział w rozwoju stanu zapalnego skóry. 16, 17 W niniejszym artykule przedstawimy dylematy dotyczące etiopatogenezy atopowego zapalenia skóry, z uwzględnieniem genetycznie uwarunkowanych oraz nabytych przyczyn zaburzeń struktury i funkcji bariery naskórkowej, które odpowiedzialne są za całą naturalną historię przebiegu choroby. 18, 19, 20 Przedstawimy też dylematy kliniczne, w tym trudności w rozpoznawaniu poszczególnych fenotypów AZS, oraz dylematy terapeutyczne, w świetle możliwości działania profilaktycznego, wczesnej interwencji oraz nowoczesnej terapii indywidualnie dobieranej dla konkretnego pacjenta.
Dylematy etiopatogenetyczne
Zaburzenia w zakresie bariery naskórkowej u chorych na AZS (ryc. 1)
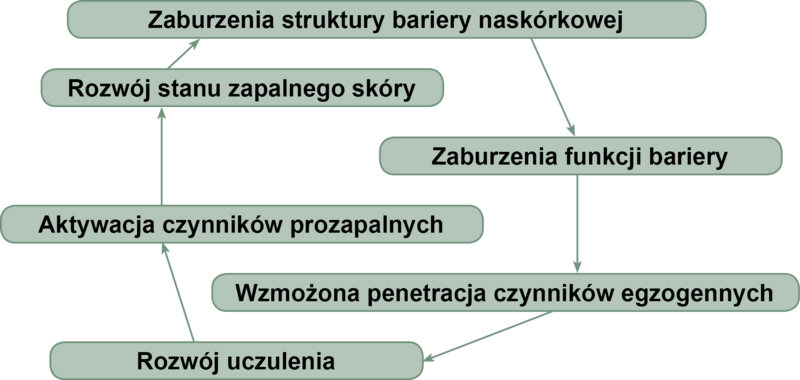
Ryc. 1. Zamknięte koło wzajemnych zależności pomiędzy barierą naskórkową i układem immunologicznym w atopowym zapaleniu skóry.
Najwięcej uwagi dotychczas poświęcono wielokierunkowej roli filagryny (FLG) w funkcjonowaniu bariery naskórkowej u chorych na AZS. 21, 22 W przypadku pacjentów, u których stwierdza się mutacje genów filagrynowych (AZS FLG), zarówno obraz kliniczny, jak i przebieg schorzenia jest odmienny w porównaniu z chorymi bez wspomnianych mutacji (AZS non-FLG). W przypadku chorych na AZS FLG nasilenie suchości skóry jest zdecydowanie bardziej zaznaczone, pierwsze objawy chorobowe występują wcześniej, sam przebieg jest bardziej przewlekły i często pojawiają się kliniczne objawy astmy, alergii na pokarmy oraz zakażeń bakteryjnych skóry. 23, 24, 25 Na podstawie obecnej wiedzy wydaje się, że wspomniane dwa typy AZS charakteryzują się odmiennymi ścieżkami rozwoju stanu zapalnego skóry. 26 W pierwszym typie rejestruje się wzmożoną ekspresję IL-1 w obrębie warstwy rogowej naskórka oraz mediowany przez interferon typ 1 odpowiedzi na stres. 27, 28 Z kolei w przypadku dzieci należących do drugiego typu (AZS non-FLG) obserwuje się zaburzenia procesów metabolizmu lipidów. 28 Wiadomo też, że zależne od filagryny uwalnianie sfingomielinazy wydaje się pełnić rolę ochronną w zakresie śmierci komórkowej indukowanej przez gronkowcową α-toksynę. 29 Obserwacje te wskazują jednoznacznie, że pacjenci wykazujący mutacje filagrynowe stanowią odmienny endotyp AZS, o zdecydowanie innym przebiegu klinicznym i możliwość ich identyfikacji mogłaby stanowić podstawy do rozwoju i wdrożenia celowanych terapii ukierunkowanych na poprawę struktury i funkcji bariery naskórkowej w tej właśnie grupie chorych.
Jak wiadomo, kliniczna manifestacja AZS uwarunkowana jest złożonymi interakcjami pomiędzy genetyką i środowiskiem oraz pomiędzy poszczególnymi genami. Genowo-środowiskowe interakcje udokumentowano badaniami na modelach mysich, które pozbawione genów filagrynowych po ekspozycji ich skóry na alergeny środowiskowe lub mikroorganizmy rozwijały objawy wyprysku. 30 Na tej samej zasadzie we wspomnianej grupie pacjentów (AZS FLG) wczesna ekspozycja na alergeny roztoczowe może prowadzić do rozwoju uczulenia na alergeny roztoczowe. 31, 32, 33
W odniesieniu do interakcji pomiędzy poszczególnymi genami w badaniach na modelach mysich (flaky tail mice) wykazano możliwość spontanicznego rozwoju klinicznych objawów AZS w sterylnych warunkach wolnych od wszelkich mikroorganizmów środowiskowych. 34, 35
W zależności od analizowanej populacji chorych na AZS mutacje FLG stwierdza się u do 40 proc. chorych o ciężkim przebiegu choroby, ale mniej niż 20 proc. w tej grupie pacjentów jest homozygotami lub złożonymi heterozygotami w odniesieniu do mutacji FLG. 36 Co więcej, mutacje FLG stwierdza się jedynie w przypadku zdecydowanej mniejszości populacji euroamerykańskiej i praktycznie nie rejestruje się ich u afroamerykańskich chorych na AZS. 21, 22, 37, 38
Wiadomo też, że istnieje wiele zróżnicowanych przyczyn ograniczających ekspresję FLG w skórze. Najczęstszą z nich jest aktywacja układu immunologicznego. 39, 40, 41 Należy jednak zdecydowanie podkreślić epigenetyczną drogę prowadzącą do tego samego efektu w odniesieniu do ekspresji FLG. 42
Dysfunkcja bariery naskórkowej niezależna od aspektu filagrynowego
Wiadome jest też, że poza elementem zależnym od FLG istnieje cały szereg innych genetycznie uwarunkowanych wariantów kodujących rozmaite białka kompleksu różnicowania naskórka zlokalizowanych w obrębie chromosomu 1q21. 43 Należą do nich m.in.:
Jednak w przeciwieństwie do FLG biologiczne znaczenie wspomnianych wariantów genowych nie jest w pełni jasne w odniesieniu do AZS. Istnieje szereg danych świadczących o istotnym znaczeniu mutacji typu „loss of function” w zakresie inhibitorów proteaz serynowych, takich jak SPINK5, czego wynikiem jest proteazozależna aktywacja ścieżek wzmacniająca odpowiedź profilu Th2. Jak widomo, ten kierunek polaryzacji immunologicznej nasila zaburzenia w zakresie struktury i funkcji bariery naskórkowej i może stymulować rozwój klinicznych objawów alergicznej reakcji w obrębie skóry. 19 Złożoność wariantów genów naskórkowych podlega następnie modyfikacji poprzez działanie kolejnych wariantów genetycznych odpowiedzialnych za kontrolę immunologicznej odpowiedzi typu wrodzonego i nabytego. 6, 24
W obrębie skóry zdrowej współdziałanie pomiędzy szerokim panelem czynników warunkujących jej szczelność powoduje prawidłowe jej nawilżenie oraz zabezpiecza przed penetracją zarówno alergenów środowiskowych, jak i rozmaitych patogenów. W sytuacji uszkodzenia tej bariery wynikającej m.in. z niedoboru strukturalnych białek takich jak filagryna, inwolukryna czy lorykryna, lipidów (np. ceramidów) lub obu tych grup wspomnieć należy o zaangażowaniu kolejnych elementów strukturalnych tworzących barierę. Należą do nich m.in. klaudyny, czyli zlokalizowane bezpośrednio pod warstwą rogową białka połączenia ścisłego (tight junction proteins), które tworzą swojego rodzaju „drugorzędową” strukturalną barierę naskórkową. 47 Profilowanie genetyczne w zakresie bariery naskórkowej u chorych na AZS wykazało ograniczenie ekspresji genów dla klaudyny i funkcji biologicznej tego białka. W sytuacji gdy obie bariery naskórkowe (filagryna i klaudyna) wykazują ewidentne zaburzenia, musi dochodzić do zapoczątkowania natychmiastowej odpowiedzi immunologicznej typu pierwotnego (wrodzonego) w celu ograniczenia inwazji patogenów środowiskowych. Jak wiadomo, zarówno keratynocyty, jak i komórki prezentujące antygen w obrębie skóry wykazują ekspresję receptorów rozpoznawania wzoru (pattern recognition receptors), takich jak receptory żetonowe (toll-like receptors – TLRs).
Pobudzenie TLRs poprzez mikroorganizmy prowadzi do uwalniania białek przeciwdrobnoustrojowych (antimicrobial peptides – AMPs) w celu wzmocnienia funkcji drugorzędowej bariery naskórkowej (ograniczenie penetracji alergenów i mikroorganizmów środowiskowych). 47 Okazuje się jednak, że w przypadku chorych na AZS funkcja TLRs jest istotnie ograniczona.
Ostatecznie ograniczenie funkcji bariery naskórkowej i nasilenie ciężkości przebiegu klinicznego AZS w zdecydowany sposób predysponuje do kolonizacji skóry chorych przez drobnoustroje i rozwoju przewlekłego przebiegu stanu zapalnego. Jest to m.in. wynikiem zwiększonej ekspresji tkankowych receptorów dla Staphylococcus aureus, a zatem kolonizacja ta jest zjawiskiem powszechnym. 48, 49 Okazuje się, że keratynocyty chorych na AZS charakteryzuje ograniczona w stopniu znacznym zdolność syntezy białek przeciwdrobnoustrojowych koniecznych do kontroli replikacji S. aureus oraz niektórych wirusów. 50, 51 Co więcej, organizmy S. aureus syntetyzują i uwalniają proteazy serynowe, które – jak już wspomniano wcześniej – prowadzą do degradacji bariery naskórkowej. 52 A zatem w przypadku źle kontrolowanego AZS funkcja bariery naskórkowej ulegać może stałej, pogłębiającej się dysfunkcji na drodze wielu zróżnicowanych mechanizmów.
Jest to dylemat, z którym nadal borykamy się w naszej naukowej i klinicznej sferze rozważań nad problemem zrozumienia i możliwej kontroli objawów AZS.
Problem immunologicznych zaburzeń w obrębie bariery naskórkowej w AZS
Mimo wielu twardych danych dotyczących znaczenia genetycznie uwarunkowanych zaburzeń strukturalnych bariery naskórkowej w AZS 18, 19 istnieje ogromna liczba równie przekonujących dowodów na dominujące znaczenie zaburzeń immunologicznych, czyli immunologicznie mediowanej polaryzacji ścieżek rozwoju stanu zapalnego skóry prowadzących do wtórnego uszkodzenia struktury i funkcji bariery naskórkowej. Oto przykłady:
1. Mutacji genów dla FLG nie stwierdza się u większości chorych na AZS. 21, 22, 53
2. Większość dzieci „wyrasta” z AZS pomimo stwierdzanych mutacji FLG. 54
3. W przeciwieństwie do dzieci chorych na rybią łuskę objawy AZS pojawiają się w zdecydowanie późniejszym okresie ich życia i dodatkowo stwierdza się objawy skóry zarówno chorej, jak i pozornie zdrowej, czyli niezmienionej chorobowo.
4. Zarówno w obrębie skóry chorej, jak i pozornie zdrowej stwierdza się szeroki zakres zaburzeń w zakresie białek różnicowania końcowego korneocytów, poza problemem filagrynowym (np. lorykryna, inwolukryna, korneodezmozyna czy klaudyny). Sugeruje to reaktywne zaburzenia w zakresie różnicowania/rogowacenia komórek naskórka. 55, 56
5. Ekspresja filagryny może być istotnie ograniczona poprzez działanie takich cytokin jak IL-4, IL-13, IL-22, IL-25 oraz IL-31, co oczywiście prowadzi w prostej linii do zaburzeń w zakresie bariery naskórkowej. 17, 19, 39, 40, 41, 56 IL-22 bezpośrednio powoduje hiperplazję keratynocytów oraz ogranicza ekspresję FLG.
6. Badania prowadzone na modelach mysich genetycznie spolaryzowanych w kierunku profilu cytokinowego typu Th2 spontanicznie rozwijają objawy kliniczne AZS oraz cechy zaburzeń bariery naskórkowej. 57, 58, 59, 60
7. Poprzez stosowanie zarówno glikokortykosteroidów, jak i inhibitorów kalcyneuryny jesteśmy w stanie wzmocnić ekspresję FLG. 61
8. Najsilniejszym argumentem wydaje się fakt, że poprawa, a czasem nawet uzyskanie stanu bezobjawowego w przebiegu AZS o średniociężkim i ciężkim nasileniu objawów klinicznych może wystąpić w efekcie zastosowania terapii immunosupresyjnej (cyklosporyna, fototerapia), 62, 63 a leki biologiczne takie jak dupilumab prowadzą do zdecydowanej poprawy w zakresie zaburzeń bariery naskórkowej w AZS.
Podkreślenia wymaga fakt, że w przypadku zapalnych zmian skórnych w AZS aktywacja immunologiczna obecna jest praktycznie zawsze. 61, 64, 65 U chorych o przewlekłym przebiegu AZS niezmiennie obecnymi cechami o charakterze immunologicznym są:
- wyraźny naciek skóry złożony z limfocytów T (przeciętnie 10-krotnie większy w porównaniu ze skórą zdrową),
- naciek mieloidalnych komórek dendrytycznych (CD11+) (również zazwyczaj 10-krotnie przewyższający obserwowany w obrębie skóry zdrowej, a komórki te mają w większości „fenotyp zapalny” (BDCA1-/CD11c+), 66
- zwiększona synteza i uwalnianie cytokin i chemokin prozapalnych przez pobudzone limfocyty T oraz komórki dendrytyczne w obrębie zmian skórnych,
- reaktywna hiperplazja naskórka lub „dojrzewanie regeneracyjne”, wykazujące nietypową odpowiedź typu hiperplazji, w której mRNA oraz białka rogowacenia naskórkowego są w wysokim stopniu hamowane poprzez keratynocyty objętego procesem chorobowym obszaru skóry atopowej. 17, 67, 68, 69
Immunologiczne ścieżki rozwoju zmian skórnych u chorych na AZS
W przypadku chorych na AZS o wysokich stężeniach surowiczej IgE w obrębie skóry niezmienionej chorobowo obserwuje się selektywną ekspansję limfocytów Th2 o lokalizacji okołonaczyniowej (tabela 2). 64 Ostre, naciekowe zmiany skórne charakteryzuje aktywacja komórek Th2, Th22 oraz Th17, czego wyrazem jest synteza i uwalnianie cytokin prozapalnych, typowych dla tych linii komórkowych. Równocześnie dochodzi do rozwoju odpowiedzi naskórkowej (S100), wyrażonej wybitnie wzmocnioną ekspresją genów S100A (S100A7-9), które są częścią prozapalnego kompleksu różnicowania naskórka, co jak wiadomo, podlega regulacji cytokinowej (IL-22 oraz IL-17). 74 Natomiast w przypadku chorych o przewlekłym przebiegu AZS aktywacja typu Th2 i Th22 przebiega równocześnie z istotnym elementem Th1-zależnym, przy czym nie obserwuje się „kompletnego przełączenia” w kierunku Th1. 17 Pomimo że profil cytokinowy typu Th2 rejestrowany jest nadal w fazie przewlekłego stanu zapalnego w AZS, typowy jest wzrost stężenia IFN-γ odpowiedzialnego m.in. za apoptozę keratynocytów. 70

Tabela 2. Wzajemne zależności pomiędzy zaburzeniami immunologicznymi oraz strukturalnymi w obrębie bariery naskórkowej u chorych na AZS
Na przestrzeni ostatniej dekady stwierdzono, że cytokiny Th2- i Th22-zależne w istotnym zakresie modulują barierę naskórkową w AZS, w tym ograniczają różnicowanie keratynocytów, hiperplazję, apoptozę keratynocytów oraz syntezę białek przeciwdrobnoustrojowych. 39, 40, 41, 50, 71, 72, 73, 74
Do działań niepożądanych cytokin należą:
- hamowanie genów dla białek końcowego różnicowania keratynocytów (np. FLG, lorykryna, inwolukryna) poprzez cytokiny profilu Th2 (IL-4, IL-13 oraz IL-31) oraz cytokiny profilu Th22/IL-22,
- hamowanie syntezy białek przeciwdrobnoustrojowych przez cytokiny profilu Th2 (IL-4, IL-13),
- wzmocnienie S100As przez IL-17 i IL-22,
- wzmocnienie hiperplazji naskórka przez cytokiny profilu Th22/IL-22. 69
Aktywność procesu chorobowego u pacjentów z AZS (ocena w skali SCORAD) pozostaje w dodatniej korelacji z ekspresją mediatorów o charakterze Th2 oraz Th22 w obrębie zapalnych zmian skórnych oraz w zależności ujemnej z markerami różnicowania końcowego keratynocytów. 62, 71, 72, 73, 74 Pomimo że aktywacja immunologiczna w obrębie zapalnych zmian skórnych jest istotnie wyższa w porównaniu ze skórą niezmienioną w AZS, ekspresja szerokiego zakresu genów różnicowania naskórkowego (czyli lorykryna, periplakina oraz inwolukryna wraz z filagryną) charakteryzuje zarówno skórę objętą procesem chorobowym, jak i pozornie zdrową. Ponieważ cytokiny profilu Th2 (IL-4/IL-13), jak też Th22 (IL-22) mają zdolność hamowania ekspresji produktów końcowego różnicowania keratynocytów, podwyższone stężenie krążących cytokin o wspomnianym profilu może warunkować ograniczenie ekspresji białek bariery naskórkowej, jak też podwyższać stężenie S10As, które obserwowane jest w momencie rozwoju ostrych, zapalnych zmian skórnych. 17
Zatem nadal pozostaje wiele elementów niejasnych i co prawda możliwych do logicznej interpretacji, ale dylematem pozostaje pytanie, czy interpretacja ta jest faktycznie prawidłowa.
Abstract
ABSTRACT
Atopic dermatitis (AD) is the most common chronic inflammatory skin disease. It often precedes the development of food allergy and asthma.
Recent insights into AD reveal abnormalities in terminal differentiation of the epidermal epithelium leading to a defective stratum corneum that allows enhanced allergen penetration and systemic IgE-sensitization. Atopic skin is also predisposed to colonization or infection by pathogenic microbes, most notably Staphylococcus aureus and herpes simplex virus. The causes of this abnormal skin barrier in AD are complex and driven by a combination of genetic, environmental and immunologic factors. These factors likely account for heterogeneity of AD onset, severity and natural history of the disease.
Recent studies suggest that prevention of AD can be achieved through early intervention to protect the skin barrier. Onset of lesional AD requires effective control of local and systemic immune activation for optimal therapeutic strategy. Early intervention might improve long-term outcomes for AD and reduce the systemic allergen sensitization that leads to associated allergic diseases in the gastrointestinal and respiratory tract.
KEYWORDS: atopic dermatitis, eczema, epidermis, immune system, infection, filaggrin.
- 1. Shaw TE, Currie GP, Koudelka CW, Simpson EL. Eczema prevalence in the United States: data from the 2003 National Survey of Children’s Health. J Invest Dermatol 2011;131:67-73
- 2. Schmitt J, Langan S, Deckert S, Svensson A, von Kobyletzki L, Thomas K, et al. Assessment of clinical signs of atopic dermatitis: a systematic review and recommendation. J Allergy Clin Immunol 2013;132:1337-47
- 3. Yaghmaie P, Koudelka CW, Simpson EL. Mental health comorbidity in patients with atopic dermatitis. J Allergy Clin Immunol 2013;131:428-33
- 4. Boguniewicz M, Leung DYM. Atopic dermatitis: a disease of altered skin barrier and immune dysregulation. Immunol Rev 2011;242:233-46
- 5. McLean WH, Palmer CN, Henderson J, Kabesch M, Weidinger S, Irvine AD. Filaggrin variants confer susceptibility to asthma. J Allergy Clin Immunol 2008;121:1294-5
- 6. Barnes KC. An update on the genetics of atopic dermatitis: scratching the surface in 2009. J Allergy Clin Immunol 2010;125:16-29
- 7. Roduit C, Frei R, Loss G, Buchele G, Weber J, Depner M, et al. Development of atopic dermatitis according to age of onset and association with early-life expo-sures. J Allergy Clin Immunol 2012;130:130-6
- 8. Illi S, Depner M, Genuneit J, Horak E, Loss G, Strunz-Lehner C, et al. Protection from childhood asthma and allergy in Alpine farm environments-the GABRIEL Advanced Studies. J Allergy Clin Immunol 2012;129:1470-7
- 9. Kim J, Kim EH, Ohm I, Jung K, Han Y, Cheong HK, et al. Symptoms of atopic dermatitis are influenced by outdoor air pollution. J Allergy Clin Immunol 2013;132:495-8
- 10. Camargo CA Jr, Ganma D, Sidbury R, Erdenedelger KH, Radnaakhand N, Khandsuren B. Randomized trial of vitamin D supplementation for winter-related atopic dermatitis in children. J Allergy Clin Immunol 2014;134:831-5
- 11. Garmhausen D, Hagemann T, Bieber T, Dimitriou I, Fimmers R, Diepgen T, et al. Characterization of different courses of atopic dermatitis in adolescent and adult patients. Allergy 2013;68:498-506
- 12. Bieber T. Atopic dermatitis 2.0: from the clinical phenotype to the molecular taxonomy and stratified medicine. Allergy 2012;67:1475-82
- 13. Ingram JL, Kraft M. IL-13 in asthma and allergic disease: asthma phenotypes and targeted therapies.
- 14. Carolan BJ, Sutherland ER. Clinical phenotypes of chronic obstructive pulmonary disease and asthma: recent advances. J Allergy Clin Immunol 2013;131:627-34
- 15. Akdis CA, Bachert C, Cingi C, Dykewicz MS, Hellings PW, Naclerio RM, et al. Endotypes and phenotypes of chronic rhinosinusitis: a PRACTALL document of the European Academy of Allergy and Clinical Immunology and the American Academy of Allergy, Asthma & Immunology. J Allergy Clin Immunol 2013;131:1479-90
- 16. Beck LA, Thaci D, Hamilton JD, Graham NM, Bieber T, Rocklin R, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N Engl J Med 2014;371:130-9
- 17. Gittler J, Shemer A, Suárez-Fariñas M, Fuentes-Duculan J, Gulewicz KJ, Wang CQF, et al. Progressive activation of T(H)2/T(H)22 cytokines and selectiveepidermal proteins characterizes acute and chronic atopic dermatitis. J Allergy Clin Immunol 2012;130:1344-54
- 18. Elias P, Wakefield JS. Mechanisms of abnormal lamellar body secretion and the dysfunctional skin barrier in patients with atopic dermatitis. J Allergy Clin Immunol 2014;134:781-91
- 19. Samuelov L, Sprecher E. Peeling off the genetics of atopic dermatitis-like congenital disorders. J Allergy Clin Immunol 2014;134:808-15
- 20. Thyssen JP, Kezic S. Causes of epidermal filaggrin reduction and their role in the pathogenesis of atopic dermatitis. J Allergy Clin Immunol 2014;134:792-9
- 21. McAleer MA, Irvine AD. The multifunctional role of filaggrin in allergic skin disease. J Allergy Clin Immunol 2013;131:280-91
- 22. Irvine AD, McLean WHI, Leung DYM. Filaggrin mutations associated with skin and allergic diseases. N Engl J Med 2011;365:1315-27
- 23. Margolis DV, Apter AJ, Gupta J, Hoffstad O, Papadopoulos M, Campbell LE et al. The persistence of atopic dermatitis and filaggrin (FLG) mutations in a US longitudinal cohort. J Allergy Clin Immunol 2012;130:912-7
- 24. Bohme M, Soderholl C, Kull I, Bergstrom A, van Hage M, Wahlgren C. Filaggrin mutations increase the risk for persistent dry skin and eczema independent of sensitization. J Allergy Clin Immunol 2012;129:1153-5
- 25. Kawasaki H, Nagao K, Kubo A, Hata T, Shimizu A, Mizuno H, et al. Altered stratum corneum barrier and enhanced percutaneous immune responses in filaggrin-null mice. J Allergy Clin Immunol 2012;129:1538-46
- 26. McAleer MA, Irvine AD. The multifunctional role of filaggrin in allergic skin disease. J Allergy Clin Immunol 2013;131:280-91
- 27. Kezic S, O’Regan GM, Lutter R, Jakasa I, Koster ES, Saunders S, et al. Filaggrin loss-of-function mutations are associated with enhanced expression of IL-1 cytokines in the stratum corneum of patients with atopic dermatitis and in a murine model of filaggrin deficiency. J Allergy Clin Immunol 2012;129:1031-9
- 28. Cole C, Kroboth K, Schurch NJ, Sandilands A, Sherstnev A, O’Regan GM, et al. Filaggrin-stratified transcriptome analysis of paediatric skin identifies mechanistic pathways in atopic dermatitis. J Allergy Clin Immunol 2014;134:82-91
- 29. Brauweiler AM, Bin L, Kim BE, Oyoshi MK, Geha RS, Goleva E, et al. Filaggrin dependent secretion of sphingomyelinase protects against Staphylococcal alphatoxin-induced keratinocyte death. J Allergy Clin Immunol 2013;131:421-7
- 30. Oyoshi MK, Murphy GF, Geha RS. Filaggrin-deficient mice exhibit Th17-dominated skin inflammation and permissiveness to epicutaneous sensitization with protein antigen. J Allergy Clin Immunol 2009;124:485-93
- 31. Venkataraman D, Soto-Ramirez N, Kurukulaaratcy RJ, Holloway JW, Karmaus W, Ewart SL, et al. Filaggrin loss-of-function mutations are associated with food allergy in childhood and adolescence. J Allergy Clin Immunol 2014;134:876-82
- 32. Brough HA, Simpson A, Makinson K, Hankinson J, Brown S, Douiri A, et al. Peanut allergy: effect of environmental peanut exposure in children with filaggrin loss-of-function mutations. J Allergy Clin Immunol 2014;134:867-75
- 33. Brough HA, Liu AH, Sicherer S, Makinson K, Douiri A, Brown S, et al. Atopic dermatitis increases the impact of exposure to peanut antigen in dust on peanut sensitization and likely peanut allergy. J Allergy Clin Immunol 2014; In press
- 34. Saunders SP, Goh CS, Brown SJ, Palmer CN, Porter RM, Cole C, et al. Tmem79/Matt is the matted mouse gene and is a predisposing gene for atopic dermatitis in human subjects. J Allergy Clin Immunol 2013;132:1121-9
- 35. Sasaki T, Shiohama A, Kubo A, Kawasaki H, Ishida-Yamamoto A, Yamada T et al. A homozygous nonsense mutation in the gene for Tmem79, a component for lamellar granule secretory system, produces spontaneous eczema in an exper- imental model of atopic dermatitis. J Allergy Clin Immunol 2013;132:1111-20
- 36. Mohiuddin M, Ramamoorthy P, Reynolds PR, Curran-Everett D, Leung DYM. Increased compound heterozygous filaggrin mutations in severe atopic dermatitis in the United States. J Allergy Clin Immunol Pract 2013;1:534-6
- 37. Thawer-Esmail F, Jakasa I, Todd G, Wen YR, Brown SJ, Kroboth K, et al. South African amaXhosa patients with atopic dermatitis have decreased levels of filag- grin breakdown products but no loss-of-function mutations in filaggrin. J Allergy Clin Immunol 2014;133:280-2
- 38. Garrett JPD, Hoffstad O, Apter AJ, Margolis DJ. Racial comparison of filaggrin null mutations in asthmatic patients with atopic dermatitis in a US population. J Allergy Clin Immunol 2013;132:1232-4
- 39. Howell MD, Kim BE, Gao P, Grant AV, Boguniewicz M, DeBenedetto A, et al. Cytokine modulation of atopic dermatitis filaggrin skin expression. J Allergy Clin Immunol 2007;120:150-5
- 40. Kim BE, Bin L, Ye YM, Ramamoorthy P, Leung DYM. IL-25 Enhances HSV-1 replication by inhibiting filaggrin expression, and acts synergistically with TH2 cytokines to enhance HSV-1 replication. J Invest Dermatol 2013;133:2678-85
- 41. Cornelissen C, Marquardt Y, Czaja K, Wenzel J, Luscher-Firzlaff J, Luscher B, et al. IL-31 regulates differentiation and filaggrin expression in human organotypic skin models. J Allergy Clin Immunol 2012;129:426-33
- 42. Rodriguez E, Baurecht H, Wahn AF, Kretschmer A, Hotze M, Zeilinger S, et al. An integrated epigenetic and transcriptomic analysis reveals distinct tissue- specific patterns of DNA methylation associated with atopic dermatitis. J Invest Dermatol 2014;134:1873-83
- 43. Pellerin L, Henry J, Hsu CY, Balica S, Jean-Decoster C, Mechin MC, et al. Defects of filaggrin-like proteins in both lesional and nonlesional atopic skin. J Allergy Clin Immunol 2013;131:1094-102
- 44. Margolis DJ, Gupta J, Apter AJ, Ganguly T, Hoffstad O, Papadopoulos M, et al. Filaggrin-2 variation is associated with more persistent atopic dermatitis in African American subjects. J Allergy Clin Immunol 2014;133:784-9
- 45. Henry J, Hsu CY, Haftek M, Nachat R, de Koning HD, Gardinal-Galera I, et al. Hornerin is a component of the epidermal cornified cell envelopes. FASEB J 2011;25:1567-76
- 46. Marenholz I, Rivera VA, Esparza-Gordillo J, Bauerfeind A, Lee-Kirsch MA, Ciechanowicz A, et al. Association screening in the Epidermal Differentiation Complex (EDC) identifies an SPRR3 repeat number variant as a risk factor for eczema. J Invest Dermatol 2011;131:1644-9
- 47. Kuo I, Yoshida T, De Benedetto A, Beck LA. The cutaneous innate immune response in patients with atopic dermatitis. J Allergy Clin Immunol 2013;131:266-78
- 48. Cho SH, Strickland I, Tomkinson A, Gelfand EW, Leung DYM. Preferential binding of Staphylococcus aureus to skin sites of Th2-mediated inflammation in a murine model. J Invest Dermatol 2001;116:658-63
- 49. Cho SH, Strickland I, Boguniewicz M, Leung DYM. Fibronectin and fibrinogen contribute to the enhanced binding of Staphylococcus aureus to atopic skin. J Allergy Clin Immunol 2001;108:269-74
- 50. Ong PY, Ohtake T, Brandt C, Strickland I, Boguniewicz M, Ganz T, et al. Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N Engl J Med 2002;347:1151-60
- 51. Howell MD, Wollenberg A, Gallo RL, Flaig M, Streib JE, Wong C, et al. Cathelicidin deficiency predisposes to eczema herpeticum. J Allergy Clin Immunol 2006;117:836-41
- 52. Schlievert PM, Strandberg KL, Lin YC, Peterson ML, Leung DYM. Secreted virulence factor comparison between methicillin-resistant and methicillin-sensitive Staphylococcus aureus, and its relevance to atopic dermatitis. J Allergy Clin Immunol 2010;125:39-49
- 53. Czarnowicki T, Krueger JG, Guttman-Yassky E. Skin barrier and immune dysregulation in atopic dermatitis: an evolving story with important clinical implications. J Allergy Clin Immunol Pract 2014;2:371-9
- 54. Henderson J, Northstone K, Lee SP, Liao H, Zhao Y, Pembrey M, et al. The burden of disease associated with filaggrin mutations: a population-based longitudinal birth cohort study. J Allergy Clin Immunol 2008;121:872-7
- 55. Guttman-Yassky E, Suarez-Farinas M, Chiricozzi A, Nograles KE, Shemer A, Fuentes-Duculan J, et al. Broad defects in epidermal cornification in atopic dermatitis identified through genomic analysis. J Allergy Clin Immunol 2009;124:1235-44
- 56. Suarez-Farinas M, Tintle SJ, Shemer A, Chiricozzi A, Nograles K, Cardinale I, et al. Nonlesional atopic dermatitis skin is characterized by broad terminal differentiation defects and variable immune abnormalities. J Allergy Clin Immunol 2011;127:954-64
- 57. Chan LS, Robinson N, Xu L. Expression of interleukin-4 in the epidermis of transgenic mice results in a pruritic inflammatory skin disease: an experimental animal model to study atopic dermatitis. J Invest Dermatol 2001;117:977-83
- 58. Sehra S, Yao Y, Howell MD, Nguyen ET, Kansas GS, Leung DYM, et al. IL-4 regulates skin homeostasis and the predisposition towards allergic skin inflammation. J Immunol 2010;184:3186-90
- 59. Dillon SR, Sprecher C, Hammond A, Bilsborough J, Rosenfeld-Franklin M, Presnell SR, et al. Interleukin 31, a cytokine produced by activated T cells, induces dermatitis in mice. Nat Immunol 2004;5:752-60
- 60. Ziegler SF. Thymic stromal lymphopoietin and allergic disease. J Allergy Clin Immunol 2012;130:845-52
- 61. Jensen JM, Pfeiffer S, Witt M, Brautigam M, Neumann C, Weichenthal M, et al. Different effects of pimecrolimus and betamethasone on the skin barrier in patients with atopic dermatitis. J Allergy Clin Immunol 2009;124:19-28
- 62. Tintle S, Shemer A, Suarez-Farinas M, Fujita H, Gilleaudeau P, Sullivan-Whalen M, et al. Reversal of atopic dermatitis with narrow-band UVB phototherapy and biomarkers for therapeutic response. J Allergy Clin Immunol 2011;128:583-93
- 63. Khattri S, Shemer A, Rozenblit M, Dhingra N, Czarnowicki T, Finney R, et al. Cyclosporine in patients with atopic dermatitis modulates activated inflammatory pathways and reverses epidermal pathology. J Allergy Clin Immunol 2014;133:1626-3.
- 64. Leung DYM, Bhan AK, Schneeberger EE, Geha RS. Characterization of the mononuclear cell infiltrate in atopic dermatitis using monoclonal antibodies. J Allergy Clin Immunol 1983;71:47-56
- 65. Hamid Q, Boguniewicz M, Leung DYM. Differential in situ cytokine gene expression in acute versus chronic atopic dermatitis. J Clin Invest 1994;94:870-6
- 66. Novak N. An update on the role of human dendritic cells in patients with atopic dermatitis. J Allergy Clin Immunol 2012;129:879-86
- 67. Dhingra N, Suarez-Farinas M, Fuentes-Duculan J, Gittler JK, Shemer A, Raz A, et al. Attenuated neutrophil axis in atopic dermatitis compared to psoriasis reflects T(H)17 pathway differences between these diseases. J Allergy Clin Immunol 2013;132:498-501
- 68. Boyman O, Werfel T, Akdis CA. The suppressive role of IL-10 in contact and atopic dermatitis. J Allergy Clin Immunol 2012;129:160-1
- 69. Gittler JK, Krueger JG, Guttman-Yassky E. Atopic dermatitis results in intrinsic barrier and immune abnormalities: implications for contact dermatitis. J Allergy Clin Immunol 2013;131:300-13
- 70. Goyette J, Geczy CL. Inflammation-associated S100 proteins: new mechanisms that regulate function. Amino Acids 2011;41:821-42
- 71. Rebane A, Zimmermann M, Aab A, Baurecht H, Koreck A, Karelson M, et al. Mechanisms of IFN-induced apoptosis of human skin keratinocytes in patientswith atopic dermatitis. J Allergy Clin Immunol 2012;129:1297-306
- 72. Howell MD, Fairchild HR, Kim BE, Bin L, Boguniewicz M, Redzic JS, et al. Th2 cytokines act on S100/A11 to downregulate keratinocyte differentiation. J Invest Dermatol 2008;128:2248-58
- 73. Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R. IL-22 increases the innate immunity of tissues. Immunity 2004;21:241-54
- 74. Wolk K, Witte E, Wallace E, Docke WD, Kunz S, Asadullah K, et al. IL-22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratinocytes: a potential role in psoriasis. Eur J Immunol 2006;36:1309-23
Następny artykuł:


 Dodaj do ulubionych
Dodaj do ulubionych
