



Co znajdziesz w artykule?
- Szczegółowe omówienie rzadkich mikroangiopatii związanych z mutacjami w genach COL4A1 i COL4A2, NOTCH3, GLA, HTRA1 oraz TREX1 będących przyczynami udarów u osób młodych i udarów o niejasnej etiologii
- Przy niejednoznacznym obrazie radiologicznym udaru konieczne jest rozszerzenie diagnostyki, zwłaszcza jeśli dotyczy on osób młodych bez ewidentnego wywiadu nadciśnienia tętniczego i innych czynników ryzyka, a wywiad rodzinny jest obciążający
Spis treści
W ostatnich latach coraz większą uwagę zwraca się na udary mózgu u osób młodych i udary o niejasnej etiologii. Jednymi z możliwych przyczyn są choroby małych naczyń (SVD – small vessel disease). Te przewlekłe i postępujące schorzenia dotyczą drobnych, przeszywających naczyń krwionośnych mózgu, tzn. małych tętnic, tętniczek, naczyń włosowatych i małych żył zaopatrujących istotę białą i głębokie struktury istoty szarej 1 .
Choroby małych naczyń są związane z mutacjami w genach COL4A1 i COL4A2
, NOTCH3, GLA, HTRA1 oraz TREX1. Są to rzadkie schorzenia objawiające się charakterystycznymi zespołami neurologicznymi, a także specyficznymi zmianami radiologicznymi oraz widocznymi w badaniach neuropatologicznych¹ 0, 1, 2, 3 . Klinicznie fenotypy choroby małych naczyń różnią się w zależności od postaci. W postaci ostrej objawiają się udarami lakunarnymi bądź głębokimi krwotokami, a w postaci przewlekłej – napadami migreny z aurą, objawami pozapiramidowymi, otępieniem, depresją czy zaburzeniami chodu (tab. 1) 3 . Ich występowaniu mogą towarzyszyć objawy nieneurologiczne (zmiany skórne, oczne, kostne, zaburzenia hormonalne, nefrologiczne i kardiologiczne).
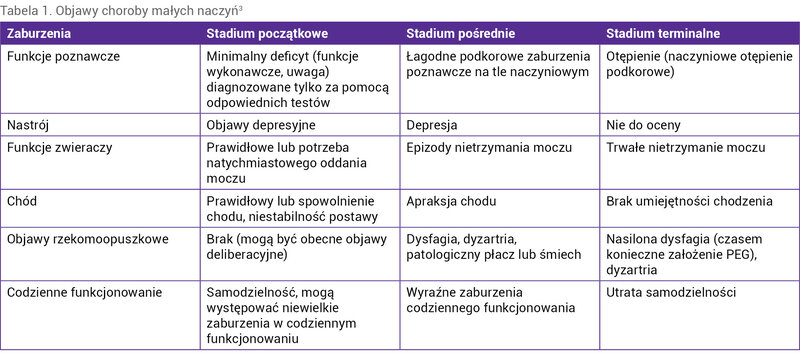
Tabela 1. Objawy choroby małych naczyń3
Przy niejednoznacznym obrazie radiologicznym konieczne jest rozszerzenie diagnostyki, zwłaszcza jeśli udar dotyczy osób młodych bez ewidentnego wywiadu nadciśnienia tętniczego i innych czynników ryzyka, a wywiad rodzinny jest obciążający. Często o ostatecznym rozpoznaniu rozstrzyga badanie genetyczne.
Zespół CADASIL
Zespół CADASIL (mózgowa autosomalna dominująca arteriopatia z podkorowymi zawałami i leukoencefalopatią; cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy) to spowodowana mutacją genu NOTCH3 na chromosomie 19p13.12 genetycznie uwarunkowana choroba małych naczyń mózgowych 4 . Szacuje się, że CADASIL występuje z częstością 1:25 000-50 000, ale dokładne dane nie są znane ze względu na trudności w rozpoznawaniu choroby. W niektórych izolowanych populacjach może występować zdecydowanie częściej niż w populacji ogólnej 3, 4, 5 . Objawy typowe dla tej choroby to migrena z aurą (20-40%), udar mózgu (40-80%) i zaburzenia poznawcze (100%) 3 . Migrena z aurą pojawia się w trzeciej dekadzie życia, udar – w czwartej, a zaburzenia poznawcze w szóstej dekadzie życia. Mogą wystąpić również zaburzenia nastroju i apatia (około 20% chorych), padaczka (10%), mikrokrwawienia (31-69%), sporadycznie – udary krwotoczne i objawy pozapiramidowe 4 . Objawy choroby występują w różnych konfiguracjach i u pacjentów w różnym wieku. Zespół ma bardzo różnorodny obraz kliniczny, nawet wśród członków tej samej rodziny 2, 3 .
Rezonans magnetyczny wykazuje bardzo charakterystyczny obraz zmian istoty białej (zmiany w sekwencji T2 wyprzedzają objawy kliniczne o około 15 lat i są widoczne już około 35 r.ż.). Zwykle umiejscowione są podkorowo i okołokomorowo, szczególnie w płacie skroniowym, torebce zewnętrznej, płatach czołowym i ciemieniowym. Zajęcie przedniej części płata skroniowego (objaw O’Sullivana) i torebki zewnętrznej uważane jest za charakterystyczną radiologiczną cechę choroby 3 .
W badaniu neuropatologicznym pacjentów cierpiących na CADASIL stwierdza się zmiany typowe dla przewlekłej choroby małych naczyń mózgu, zlokalizowane w istocie białej półkul mózgowych w okolicach okołokomorowych i jądrze półowalnym. Udary lakunarne są umiejscowione nie tylko w istocie białej, lecz także w jądrach podkorowych 2, 3 .
W badaniu histopatologicznym stwierdza się zwyrodnienie i ubytek miocytów gładkich w tętnicach małego i średniego kalibru oraz gromadzenie się w ścianach małych naczyń osmofilnego materiału ziarnistego (GOM – granular osmiophilic material) 4 . Chociaż objawy choroby wiążą się głównie z układem nerwowym, zmiany GOM obserwuje się też w innych organach: śledzionie, wątrobie, nerkach, mięśniach oraz w skórze. Dzięki łatwemu dostępowi czułość rozpoznania choroby za pomocą biopsji skóry jest wysoka 3 .
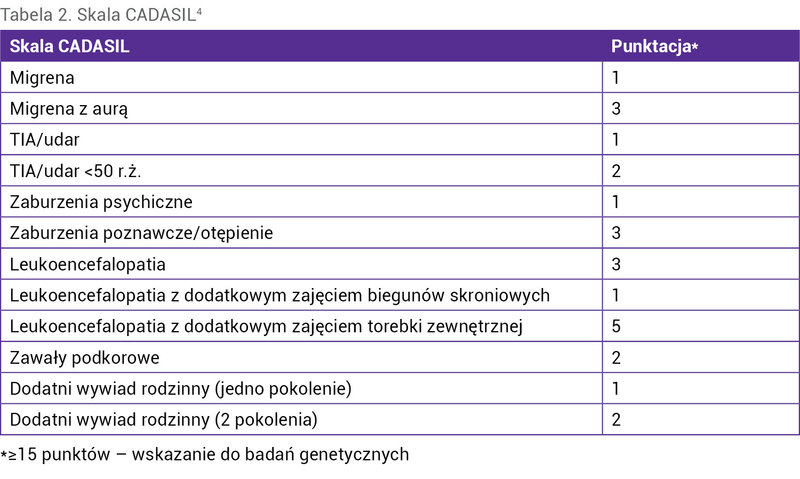
Tabela 2. Skala CADASIL4
Kryteriami umożliwiającymi rozpoznanie choroby są dodatni wynik badania genetycznego oraz obecność złogów GOM w naczyniach w biopsji skóry lub mięśnia szkieletowego. W ustaleniu wskazań do badania genetycznego może być przydatna skala CADASIL (tab. 2) 4 . Ze względu na nieznany patomechanizm choroby możliwe jest tylko leczenie objawowe, podobne jak w migrenie sporadycznej czy udarze mózgu.
Zespół CARASIL
Podobnym do zespołu CADASIL schorzeniem, również związanym z uszkodzeniem małych naczyń, jest zespół CARASIL (autosomalna recesywna arteriopatia z zawałami podkorowymi i leukoencefalopatią; cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy) występujący głównie w Azji i zwany przez autorów japońskich zespołem Maeda 6, 7 .
Zespół CARASIL wywołany jest mutacją w genie HTRA1 na chromosomie 10q26.13, kodującym peptydazę serynową 1 HtrA, która hamuje przekazywanie sygnałów przez działanie na białka należące do rodziny transformujących czynników wzrostu β 7 . Częstość jego występowania nie jest znana. Większość chorych pochodzi z Japonii, pozostałe przypadki o powiązaniach rodzinnych opisano w Hiszpanii i Chinach 6, 7 .
Początek choroby jest zmienny, a pierwszymi objawami są zwykle rozległe łysienie i zaburzenia chodu, które często zaczynają się przed ukończeniem 30 roku życia 7 . Napady ostrego bólu kręgosłupa w odcinku piersiowym lub lędźwiowym pojawiają się zazwyczaj między 20 a 45 r.ż. U połowy pacjentów występuje typowy udar niedokrwienny zatokowy, a u pozostałych następuje stopniowe pogorszenie czynności mózgu z postępującymi zaburzeniami poznawczymi, prowadzącymi do ciężkiego otępienia. Zaburzenia te zaczynają się ujawniać w wieku 30-40 lat. W stadiach zaawansowanych rozwijają się niestabilność emocjonalna, abulia i mutyzm akinetyczny. Chorzy są niezdolni do samodzielnej egzystencji mniej więcej po10 latach od początku zachorowania, ale mogą żyć 20-30 lat z chorobą 6, 7 .
W rezonansie magnetycznym stwierdza się rozlane zmiany istoty białej o hiperintensywnych sygnałach oraz liczne udary lakunarne w okolicy jąder podkorowych, wzgórza i pnia mózgu 7 . Dodatkowo w obrazach kręgosłupa można uwidocznić liczne przepukliny jąder miażdżystych krążków międzykręgowych.
Histopatologicznie CARASIL przejawia się stwardnieniem małych przeszywających naczyń mózgu, bez GOM 8 . Rozpoznanie ustala się na podstawie obecności charakterystycznych objawów klinicznych oraz wyników obrazowania rezonansu magnetycznego. Diagnozę potwierdza badanie genetyczne wykazujące mutację w HTRA1.
Zespół CARASAL
Zespół CARASAL (katepsynozależna arteriopatia z udarami i leukoencefalopatią; cathepsin A-related arteriopathy with strokes and leukoencephalopathy) jest chorobą małych naczyń spowodowaną mutacją genu CTSA na chromosomie 20q13.12, kodującego katepsynę A 9 . Najczęstszymi objawami są migreny z aurą oraz niedokrwienne udary mózgu. Mogą występować również krwotoki podpajęczynówkowe. Średni wiek zachorowania to 35 lat, a narastające otępienie jest bardzo nasilone już w wieku około 40 lat. Choroba jest bardzo rzadka (dotychczas opisano jedynie kilka przypadków na świecie) 9 .
O rozpoznaniu CARASAL należy myśleć u młodych pacjentów z chorobą małych naczyń oraz nasilonymi zmianami w istocie białej w wywiadzie rodzinnym, przy braku mutacji genów NOTCH3, HTRA1 i COL4A1/A2 9 . W takich przypadkach rozstrzygające jest badanie genetyczne w kierunku mutacji genu CTSA.
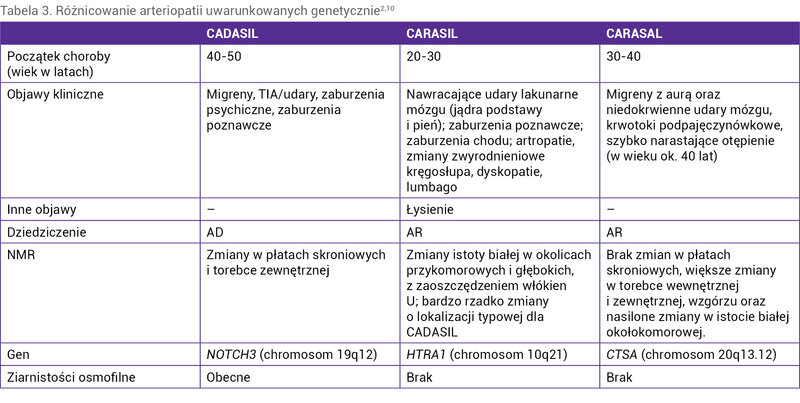
Tabela 3. Różnicowanie arteriopatii uwarunkowanych genetycznie2,10
Różnicowanie arteriopatii uwarunkowanych genetycznie przedstawiono w tabeli 3.
Choroba Fabry’ego
Choroba Fabry’ego jest lizosomalną chorobą spichrzeniową dziedziczoną z chromosomem X. Objawy występujące u kobiet związane są z przypadkową inaktywacją chromosomu X 10 . Przyczyną choroby jest mutacja genu GLA (Xq22) kodującego α-galaktozydazę A (α-Gal A; triheksozydazę ceramidową). Niedobór enzymu powoduje upośledzenie metabolizmu globotriaosylceramidu i jego gromadzenie w lizosomach komórek naczyń, komórek zwojów nerwowych, nerkach, sercu, gałkach ocznych i innych narządach 12 . Dochodzi do uszkodzenia komórek śródbłonka naczyń, mięśnia sercowego, komórek nerwowych i komórek układu autonomicznego. Częstość występowania choroby szacuje się na 1/117 000 żywych urodzeń 10 .
Choroba ma zróżnicowany obraz kliniczny. Najczęstszymi objawami są akroparestezje, zmiany skórne (angiokeratoma), białkomocz i niewydolność nerek oraz zaburzenia neurologiczne (udary mózgu, TIA) 11 . Objawy pojawiają się zazwyczaj między 6 a 9 rokiem życia. U pacjentów z łagodną postacią choroby Fabry’ego i rezydualną aktywnością α-Gal A objawy pojawiają się w wieku dorosłym i zazwyczaj ograniczone są do chorób serca i nerek 10, 11, 12 .
Przyjęto podział na trzy postacie choroby 10 :
- postać klasyczna – cechuje się wczesnym początkiem (w wieku przedszkolnym), aktywnością enzymu <1% i występowaniem wszystkich objawów klinicznych
- postać nerkowa – rozpoczyna się około 25 r.ż., aktywność enzymu wynosi >1%, dominują objawy niewydolności nerek i kardiomiopatia, nie występuje jednak uszkodzenie OUN
- postać sercowa – rozpoczyna się około 40 r.ż., aktywność enzymu wynosi >1%, dominują objawy kardiomiopatii.
W pierwszej dekadzie życia choroba objawia się bólami mięśniowymi i parestezjami kończyn, w drugiej – zaburzeniami potliwości, bólami brzucha i biegunkami. W trzeciej dekadzie często dołącza się niewydolność nerek z proteinurią, lipidurią i krwiomoczem, a w czwartej – kardiomiopatia, przerost lewego przedsionka, uszkodzenia zastawek, zaburzenia przewodzenia. Objawy neurologiczne ujawniają się w piątej dekadzie życia 11 . Udary mózgu i przemijające niedokrwienia mózgu obserwowane są 12 razy częściej niż w populacji ogólnej (średnia wieku 25-44 lata) 10 .Charakterystyczna jest predylekcja do zajęcia naczyń tylnego kręgu. Typowe są dolichoektazje (odcinkowe poszerzenia i kręty przebieg) tętnic kręgowych i podstawnej. W późniejszym czasie dołączają się zaburzenia poznawcze i pozapiramidowe, neuropatia aksonalna, szumy uszne i utrata słuchu 10, 11, 12 .
Do charakterystycznych objawów należą również gorączka nieznanego pochodzenia, zaburzenia wydzielania potu (anhydroza), angiokeratoma (ciemnoczerwone, rogowaciejące naczyniaki na skórze bioder, ud, krocza) oraz neuropatyczne bóle kończyn, szczególnie dłoni i stóp, wymagające podawania opioidów. Mogą wystąpić zaburzenia adaptacyjne i depresyjne. Najczęstszymi objawami neurologicznymi są niedowłady połowicze i objawy z tylnego kręgu krążenia mózgowego 10 .
U pacjenta, u którego objawy sugerują chorobę Fabry’ego, należy wykonać badanie stężenia α-Gal A w surowicy krwi i leukocytach. Możliwe są badania prenatalne oraz u członków rodziny chorego pozostających bez objawów, umożliwiające wczesne rozpoczęcie leczenia 13 .
Obraz MR mózgowia wykazuje cechy uszkodzenia małych naczyń pod postacią rozsianych hiperintensywnych ognisk w istocie szarej i białej płatów czołowych i ciemieniowych w obrazach T2 i FLAIR 15 . Niemal patognomoniczny jest objaw poduszki – obustronnie hiperintensywny sygnał poduszki (złogi wapnia) w tylnej części wzgórza na obrazach T1-zależnych 14 . Występuje u około 23% chorych w trzeciej dekadzie życia. Za chorobą Fabry’ego przemawiać może również poszerzenie i kręty przebieg tętnic kręgowych i tętnicy podstawnej w badaniu angio-MR. Inne zmiany są mniej swoiste i obejmują ogniska porencefaliczne po przebytych udarach oraz zmiany istoty białej – rozsiane ogniska naczyniopochodne i rozlane zmiany okołokomorowe 14 .
Należy również przeprowadzić badania czynności nerek, EKG, badania echokardiograficzne, okulistyczne w lampie szczelinowej oraz dermatologiczne 13 .
Osoby z rozpoznaniem choroby Fabry’ego powinny być poddane kompleksowej wielospecjalistycznej diagnostyce i opiece. Wczesne ustalenie rozpoznania choroby Fabry’ego umożliwia zastosowanie zastępczej terapii enzymatycznej. Obecnie dostępne są dwa preparaty: agalzydaza β i agalzydaza α 12 . Leczenie jest najskuteczniejsze w pierwszych stadiach choroby, przed uszkodzeniem narządowym. Terapia zmniejsza ból neuropatyczny i poprawia klirens kreatyniny, ale nie ma wpływu na rokowanie. Leczenie przeciwzakrzepowe również nie wpływa na rokowanie w udarze u osób z chorobą Fabry’ego. Postuluje się włączanie terapii zastępczej u chłopców od 7 roku życia pozostających bez objawów 12 . Dodatkowo stosuje się leczenie objawowe, ukierunkowane na uszkodzone narządy. Duże nadzieje wiąże się obecnie z badaniami skriningowymi noworodków, umożliwiającymi wczesne włączenie enzymatycznej terapii zastępczej 12 .
Mikroangiopatie związane z mutacjami genów COL4A1 i COL4A2
Mikroangiopatie te są wywołane mutacją w genach COL4A1 (13q34) lub COL4A2 (13q34), które kodują łańcuchy kolagenu IV, budującego błonę podstawną naczyń krwionośnych, struktur oka, kłębuszków nerkowych oraz błonę podstawną mózgu 15 .
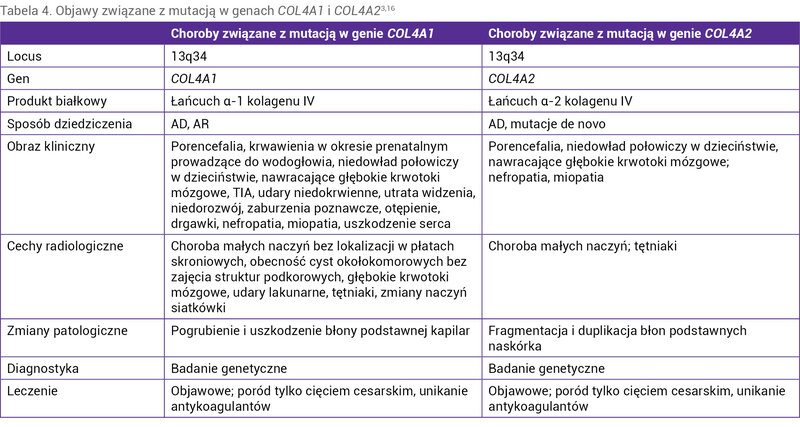
Tabela 4. Objawy związane z mutacją w genach COL4A1 i COL4A23,16
Obraz kliniczny mikroangiopatii charakteryzuje się bardzo szerokim zakresem objawów (tab. 4). Do odchyleń w badaniu neurologicznym zaliczamy niedowłady połowicze, migreny, nieprawidłowy rozwój umysłowy, zaburzenia poznawcze, otępienie, napady padaczkowe. Udary niedokrwienne i krwotoki śródmózgowe występują w każdym wieku, również w okresie prenatalnym lub w trakcie porodu. Na szczególną uwagę zasługują krwawienia, które mogą pojawiać się spontanicznie, w wyniku urazów lub leczenia przeciwkrzepliwego u pacjentów bez nadciśnienia <50 r.ż. Udar mózgu często okazuje się pierwszym objawem choroby 15 .
Wśród objawów ze strony narządu wzroku wyróżniamy jaskrę, zaćmę, przemijające niedowidzenie spowodowane krwotokami, dysgenezję przedniego odcinka gałki ocznej (anomalia Axenfelda-Riegera). Do innych częstych objawów należy zaliczyć krwiomocz, białkomocz, torbiele nerek, tętniaki tętnicy szyjnej wewnętrznej, objaw Raynauda, nadkomorowe zaburzenia rytmu i skurcze mięśniowe 15, 16, 17 .
W badaniach obrazowych charakterystyczny jest obraz podobny do angiopatii nadciśnieniowej – okołokomorowe intensywne zmiany istoty białej z udarami niedokrwiennymi, krwotocznymi, lakunarnymi i cystami okołokomorowymi 16 .
W badaniach histopatologicznych naczyń skóry stwierdza się pogrubiałą błonę podstawną naczyń włosowatych oraz fragmentację i duplikację błon podstawnych naskórka 16, 17 . Ostateczne rozpoznanie można ustalić na podstawie sekwencjonowania genów.
Dotychczas udało się wyodrębnić kilka fenotypów klinicznych zaliczanych do zakresu COL4A1:
- porencefalia dziedziczona autosomalnie dominująco typu I (autosomal dominant porencephaly type I) 16 :
- występuje w okresie noworodkowym i niemowlęcym
- charakteryzuje się widocznymi w MR jamami płynowymi (porencefalicznymi) będącymi pozostałością krwotoków śródmózgowych w życiu płodowym i noworodkowym, mikrokrwotokami, okołokomorową leukoencefalopatią, udarami lakunarnymi i zwapnieniami
- objawia się niedowładami połowiczymi u noworodków, niesprawnością intelektualną, napadami padaczkowymi, dystonią, udarami i migrenami; pierwszym objawem może być udar mózgu u wcześniej pozostającego bez objawów dorosłego
- mózgowa choroba małych naczyń mózgowych przebiegająca z krwotokiem (brain SVD with hemorrhage) 17 :
- rodzinna leukoencefalopatia naczyniowa zależna od COL4A1
- w MR charakteryzuje się zajęciem małych naczyń mózgowych z rozlaną okołokomorową leukoencefalopatią, zawałami lakunarnymi, mikrokrwawieniami, krwotokami śródmózgowymi, poszerzonymi przestrzeniami okołonaczyniowymi i zwapnieniami
- niedowłady połowicze w okresie niemowlęcym, napady padaczkowe, migreny z aurą, krwotoki śródmózgowe przed 50 r.ż., zaćma wrodzona, kręte naczynia siatkówki, anomalia przedniego odcinka oka typu Axenfelda-Riegera 18
- rzadziej występują skurcze mięśni ze wzrostem kinazy kreatynowej, uszkodzenie nerek i niedokrwistość hemolityczna
- choroba małych naczyń mózgowych z anomalią Axenfelda-Riegera (brain SVD with Axenfeld-Rieger Anomaly) 18 :
- zespół wad wrodzonych charakteryzujący się występowaniem obustronnych wad przedniego odcinka oka skojarzonych z wadami rozwojowymi zębów, środkowej części twarzy i jamy brzusznej
- dziedziczna angiopatia z krętością naczyń siatkówki, nefropatią, tętniakami i kurczami mięśni (HANAC – hereditary angiopathy with nephropathy, aneurysms, and muscle cramps syndrome) 19
- choroba małych naczyń z leukoencefalopatią, zawałami lakunarnymi, mikrokrwawieniami i poszerzonymi przestrzeniami okołonaczyniowymi, dodatkowo występują nefropatia, mnogie lub pojedyncze tętniaki tętnic szyjnych, kręte naczynia siatkówki i skurcze mięśniowe; udary niedokrwienne i krwotoczne występują rzadko
- autosomalna dominująca mikroangiopatia mostu z leukoencefalopatią (PADMAL – pontine autosomal dominant microangiopathy with leukencephalopathy) 20 :
- charakteryzuje się zawałami lakunarnymi mostu oraz uszkodzeniem podkorowej i okołokomorowej istoty białej; uszkodzenie płatów skroniowych i krwawienia występują rzadko
- nie ma leczenia przyczynowego, zapobieganie powikłaniom choroby polega na redukcji czynników ryzyka udarów mózgu (unikanie palenia tytoniu, leczenie nadciśnienia tętniczego), unikaniu leczenia przeciwkrzepliwego i urazów mogących powodować krwotoki 16 .
Zespół CRMCC
Zespół CRMCC (cerebroretinal microangiopathy with calcifications and cysts), zwany także zespołem „Coats plus”, należy do rzadkich genetycznie uwarunkowanych chorób małych naczyń 21 . Do cech charakterystycznych zespołu należą przede wszystkim zmiany wewnątrzczaszkowe w postaci zwapnień, torbieli i leukoencefalopatii, z towarzyszącym uszkodzeniem naczyń siatkówki (obecność telangiektazji i wysięków) 21 . Ponadto do objawów zespołu CRMCC należą: osteopenia z tendencją do złamań, supresja szpiku kostnego, tendencja do krwawień żołądkowo-jelitowych oraz nadciśnienie wrotne. Nazwa zespół „Coats plus” pochodzi od choroby Coatsa, w której obserwuje się izolowaną mikroangiopatię naczyń siatkówki, bez uszkodzeń ze strony innych narządów 22 . Częstość występowania zespołu szacuje się na <1/100 000.
Najczęstszym objawem zespołu CRMCC są zaburzenia ze strony OUN w postaci drgawek, ataksji, dystonii, zaburzeń ruchowych, a także deficytów poznawczych 21 . Nieprawidłowo zbudowane naczynia siatkówki predysponują do powstawania wysięków między warstwami siatkówki, co klinicznie objawia się w postaci utraty wzroku. Wraz z wiekiem dochodzi do nasilenia deficytów neurologicznych i postępującego pogorszenia widzenia. Pierwsze objawy pojawiają się we wczesnym dzieciństwie, a nawet już w życiu płodowym. Zazwyczaj obserwuje się opóźniony wzrost i nieprawidłowy rozwój kości. Na skutek osteopenii dochodzi do złamań patologicznych, a supresja szpiku kostnego powoduje niedokrwistość. Osoby z zespołem „Coats plus” charakteryzują się ponadto przedwczesnym siwieniem, nieprawidłową budową włosów i paznokci, a także zaburzoną pigmentacją skóry, mogą występować u nich plamy typu café au lait. Za zagrażające życiu powikłania zespołu uważa się marskość wątroby, nadciśnienie wrotne i poważne krwawienia żołądkowo-jelitowe 22 .
Przyczyną zespołu CRMCC jest heterozygotyczna mutacja w genie CTC1 (conserved telomere maintenance component 1) znajdującym się na chromosomie 17 (mutacja 17p13.1) 22 . Gen CTC1 koduje białko wchodzące w skład kompleksu dla replikacji telomerów, które są niezbędne dla zachowania stabilności genomu. W wyniku mutacji tego genu dochodzi do przesunięcia ramki odczytu, co prowadzi do syntezy niestabilnych bądź skróconych białek, niebędących w stanie stworzyć kompleksu z pozostałymi składnikami. W efekcie dochodzi do skrócenia telomerów bądź tworzenia chromosomów fuzalnych 22 .
Zespół RVCL
Zespół RVCL (autosomal dominant retinal vasculopathy with cerebral leukodystrophy) jest leukodystrofią mózgową z waskulopatią siatkówki dziedziczoną autosomalnie dominująco (chromosom 3p21.1 – p21.3, gen TREX1) 24 . Do zakresu choroby zaliczamy fenotypy kliniczne:
- waskulopatię mózgowo-siatkówkową (CRV – cerebro-retinal vasculopathy)
- dziedziczną endoteliopatię, retinopatię, nefropatię i udar (HERNS – hereditary endotheliopathy, retinopathy, nephropathy and stroke)
- dziedziczną retinopatię naczyniową (HVR – hereditary vascular retinopathy).
Pierwsze objawy pojawiają się w 40-50 r.ż. Zazwyczaj są to symptomy ze strony narządu wzroku o charakterze postępujących zaburzeń widzenia (waskulopatia naczyń siatkówki, nowotworzenie naczyń w tarczy nerwu wzrokowego, krwotoki do siatkówki, mikrotętniaki plamki żółtej, zanik naczyń włosowatych zaczynający się od plamki żółtej) 23 . W dalszej kolejności dołączają się objawy neurologiczne: udary niedokrwienne/TIA, migrena, zaburzenia poznawcze, zaburzenia psychiczne (zaburzenia osobowości, depresja, niepokój), drgawki, zgon w ciągu 10 lat od rozpoznania 23 . Mogą wystąpić również objawy uogólnionego niedokrwienia narządów wewnętrznych, zespół Raynauda, marskość wątroby, niewydolność nerek i martwica kości.
W MR stwierdza się rozlaną okołokomorową leukoencefalopatię i zawały lakunarne związane z zajęciem małych naczyń, czasem dodatkowo pseudotumor w postaci zmiany wzmacniającej się po środku cieniującym zlokalizowanej w mózgu lub móżdżku, otoczonej obrzękiem (podobnej do martwicy po napromieniowaniu) 23 .
Mutacje genu FOXC1 powodujące chorobę małych naczyń
Choroba jest wynikiem mutacji de novo lub mutacji dziedzicznej genu FOXC1 na chromosomie 6p25 24 . Produkt białkowy tego genu odpowiada za prawidłowy rozwój naczyń. Mutacja genu może prowadzić do udarów podkorowych i być przyczyną licznych wad (kręte naczynia siatkówki, zaćma, jaskra, dysgenezja przedniego segmentu oka, wady uzębienia, serca i pępka, zaburzenia słuchu, malformacje w móżdżku) 24 .
W MR stwierdza się rozlane zmiany istoty białej, zlokalizowane w biegunach przednich płatów skroniowych i torebce zewnętrznej, wodogłowie, heterotopię okołokomorową i malformacje naczyniowe w móżdżku 24 .
W ramach diagnostyki wykonuje się badania genetyczne, a leczenie jest tylko objawowe.
Choroba moyamoya
Chorobę moyamoya charakteryzuje zwężenie naczyń koła Willisa z powstawaniem bogatego krążenia obocznego w jądrach podstawy. Zwężenia obserwowane są w końcowych odcinkach tętnic szyjnych wewnętrznych, tętnicach środkowych i przednich mózgu. Zmiany najczęściej występują obustronnie. Słowo „moyamoya” oznacza po japońsku mgłę lub dym powstający w czasie palenia papierosów. Nazwa choroby pochodzi od charakterystycznego obrazu angiograficznego krążenia obocznego powstającego w wyniku niedrożności głównych tętnic. Choroba występuje głównie u młodych Azjatów, szczególnie w populacji japońskiej. Częściej dotyka kobiet niż mężczyzn (3:2) i w większości przypadków dotyczy dzieci. Określono dwa piki występowania objawów – między 10 a 14 i około 40 roku życia. Dotychczas opisano rodzinne występowanie choroby w 10% przypadków, z czego 76% dotyczyło rodzeństwa, a 26% rodziców i dzieci.
Choroba najprawdopodobniej dziedziczona jest autosomalnie, ale ma wieloczynnikowe podłoże. Dotychczas określono jej związek z genami: RNF213 (17q25), ACTA2 (10q23), GUCY1A3 (4q32) oraz z locus 8q23 i 3p. Występowanie choroby powiązano również z antygenem HLA-B21 i podwyższonym stężeniem przeciwciał przeciwtarczycowych 25, 26 .
W badaniach histopatologicznych naczyń obserwowane są włóknisto-komórkowe pogrubienie błony wewnętrznej, proliferacja komórek mięśni gładkich, zwiększenie ilości elastyny i gromadzenie lipidów, co prowadzi do zwężenia naczyń i powstawania krążenia obocznego.
Choroba w wieku dziecięcym objawia się głównie nagłymi niedowładami połowiczymi o podłożu niedokrwiennym i napadami padaczkowymi. U dorosłych dominują krwotoki podpajęczynówkowe. Dodatkowo występują epizody przemijającego niedokrwienia mózgu, afazja, bóle i zawroty głowy, ruchy mimowolne oraz zaburzenia poznawcze.
Złotym standardem diagnostyki jest badanie angiograficzne tętnic mózgowia. Widoczne są w nim charakterystyczne „kłęby dymu”, czyli liczne drobne naczynia krążenia obocznego powstające w wyniku niedrożności głównych tętnic. Standardowe badania MR, angio-MR, TK i dopplerowskie USG mogą być pomocne i naprowadzić na rozpoznanie. W badaniach MR i TK widoczne są zmiany niedokrwienne i krwotoczne, również przed objawową fazą choroby 27 .
W leczeniu stosuje się leki przeciwpłytkowe i rozszerzające naczynia. Podawanie antykoagulantów jest przeciwwskazane ze względu na zwiększenie ryzyka krwawień śródczaszkowych. Podstawą leczenia są chirurgiczne metody rewaskularyzacyjne 28 .
Podsumowanie
Mikroangiopatie wrodzone, mimo że są rzadkimi chorobami ośrodkowego układu nerwowego, stanowią coraz częściej rozpoznawaną przyczynę objawów naczyniowego uszkodzenia mózgu. Ich patofizjologia jest jeszcze mało poznana (z wyjątkiem zespołu CADASIL). Charakteryzują się typowym fenotypem choroby małych naczyń. Są odpowiedzialne za udary w młodym wieku i wiele przypadków udarów o nieznanej etiologii. Obok incydentów naczyniowych charakterystyczna jest obecność innych objawów neurologicznych: migreny z aurą, padaczki, zaburzeń psychicznych i zaburzeń poznawczych o wczesnym początku. Wiele z tych chorób współistnieje z uszkodzeniami siatkówki, nerek, skóry i mięśni, natomiast brakuje typowych naczyniowych czynników ryzyka, takich jak cukrzyca, nadciśnienie tętnicze, palenie papierosów. Niezmiernie ważny jest wywiad rodzinny w kierunku udarów o wczesnym początku i otępienia. W rozpoznaniu pomocne jest badanie metodą rezonansu magnetycznego, ponieważ w niektórych przypadkach zmiany radiologiczne są bardzo charakterystyczne, w większości jednak konieczne jest badanie genetyczne.
Abstract
Rare diseases of the central nervous system associated with brain vessels involvement
Ischemic stroke is most often observed in the elderly, however, it can also occur at a young age. One of the possible causes are rare genetic diseases underlying so-called small vessel disease (SVD). Congenital microangiopathies are an increasingly recognized cause of the symptoms of vascular brain damage. In addition to vascular incidents, there are other characteristic neurological symptoms: migraine with aura, epilepsy, psychiatric conditions and early-onset cognitive disorders. They may be accompanied by non-neurological symptoms (skin, eye, bone, hormonal, nephrological and cardiac disorders). If radiological image is ambiguous, it is necessary to include additional tests and diagnostic procedures, especially in the case of stroke in young people with a positive family history, however, without a clear history of hypertension or other risk factors. Genetic test results are often the decisive factor for achieving a definitive diagnosis.
- 1. Wardlaw JM, Smith E, Biessels GJ, et al. Standards for reporting vascular changes on neuroimaging STRIVE. Lancet Neurol 2013;12:822-38
- 2. Federico A, Di Donato I, Bianchi S, et al. Hereditary cerebral small vessel diseases: A review. J Neurol Sci 2012; 322(1-2):25-30
- 3. Pantoni L. Cerebral small vessel disease: from pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol 2010;9(7):689-701
- 4. Pescini F, Nannucci S, Bertaccini B, et al. The Cerebral Autosomal-Dominant Arteriopathy With Subcortical Infarcts and Leukoencephalopathy (CADASIL) Scale: a screening tool to select patients for NOTCH3 gene analysis. Stroke 2012;43(11):2871-6
- 5. Yamamoto Y, Craggs L, Baumann M, et al. Review: molecular genetics and pathology of hereditary small vessel diseases of the brain. Neuropathol Appl Neurobiol 2011;37:94-113
- 6. Fukutake T. Cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL): from discovery to gene identification. J Stroke Cerebrovasc Dis 2011;20(2):85-93
- 7. Nozaki H, Kato T, Nihonmatsu M, et al. Distinct molecular mechanisms of HTRA1 mutants in manifesting heterozygotes with CARASIL. Neurology 2016;86: 1964-74
- 8. Tikka S, Baumann M, Siitonen M, et al. CADASIL and CARASIL. Brain Pathol 2014;24:525-544
- 9. Bugiani M, Kevelam SH, Bakels HS, et al. Cathepsin A– related arteriopathy with strokes and leukoencephalopathy (CARASAL). Neurology 2016;87:1-10
- 10. Toyooka K. Fabry disease. Curr Opin Neurol 2011;24(5):463-8
- 11. Kinoshita N, Hosomi N, Matsushima H, et al. Screening for Fabry Disease in Japanese Patients with Young-Onset Stroke by Measuring α-Galactosidase A and Globotriaosylsphingosine. J Stroke Cerebrovasc Dis 2018;27(12):3563-9
- 12. Hsu TR, Niu DM. Fabry disease: Review and experience during newborn screening. Trends Cardiovasc Med 2018;28(4):274-81
- 13. Pera J. Przegląd badań genetycznych i neuroobrazowych w diagnostyce udarów mózgu o rzadkiej etiologii. Pol Przegl Neurol 2017;13(1):1-9
- 14. Cocozza S, Russo C, Pontillo G, et al. Neuroimaging in Fabry disease: current knowledge and future directions. Insights Imaging 2018;9(6):1077-88
- 15. Meuwissen ME, Halley DJ, Smit LS, et al. The expanding phenotype of COL4A1 and COL4A2 mutations: clinical data on 13 newly identified families and a review of the literature. Genet Med 2015;17(11):843-53
- 16. Lanfranconi S, Markus HS. COL4A1 mutations as a monogenic cause of cerebral small vessel disease: a systematic review. Stroke 2010;41:513-8
- 17. Gould DB, Phalan FC, van Mil SE, et al. Role of COL4A1 in small-vessel disease and hemorrhagic stroke. N Engl J Med 2006;354:1489-96
- 18. Sibon I, Coupry I, Menegon P, et al. COL4A1 mutation in Axenfeld-Rieger anomaly with leukoencephalopathy and stroke. Ann Neurol 2007;62:177-84
- 19. Alamowitch S, Plaisier E, Favrole P, et al. Cerebrovascular disease related to COL4A1 mutations in HANAC syndrome. Neurology 2009;73:1873-82
- 20. Ding XQ, Hagel C, Ringelstein EB, et al. MRI features of pontine autosomal dominant microangiopathy and leukoencephalopathy (PADMAL). J Neuroimaging 2010;20(2):134-40
- 21. Briggs TA, Abdel-Salam GMH, Balicki M, et al. Cerebroretinal microangiopathy with calcifications and cysts (CRMCC). Am J Med Genet 2008;146A:182-90
- 22. Gu P, Chang S. Functional characterization of human CTC1 mutations reveals novel mechanisms responsible for the pathogenesis of the telomere disease Coats plus. Aging Cell 2013;12:1100-9
- 23. Stam AH, Kothari PH, Shaikh A, et al. Retinal vasculopathy with cerebral leukoencephalopathy and systemic manifestations. Brain 2016;139:2209-22
- 24. French CR, Seshadri S, Destefano A, et al. Mutation of FOX1 and PITX2 induces cerebral small-vessel disease. J Clin Invest 2014;124(11):4877-81
- 25. Fujimura M, Bang OY, Kim JS. Moyamoya Disease. Front Neurol Neurosci 2016;40:204-20
- 26. Huang S, Guo ZN, Shi M, et al. Etiology and pathogenesis of Moyamoya Disease: An update on disease prevalence. Int J Stroke 2017;12(3):246-53
- 27. Ancelet C, Boulouis G, Blauwblomme T, et al. Imaging Moya-Moya disease. Rev Neurol (Paris) 2015;171(1):45-57
- 28. Liu JJ, Steinberg GK. Direct Versus Indirect Bypass for Moyamoya Disease. Neurosurg Clin N Am 2017;28(3):361-74
Pierwszy artykuł:


 Dodaj do ulubionych
Dodaj do ulubionych
