Choroby układu pozapiramidowego
Dystonie genetycznie uwarunkowane – charakterystyka podłoża molekularnego
mgr Marta Jurek1 dr n. med. Dariusz Koziorowski2 dr n. biol. Dorota Hoffman-Zacharska1 prof. dr hab. n. med. Jerzy Bal1
STRESZCZENIE
Dystonie to heterogenna grupa chorób układu pozapiramidowego o zróżnicowanym obrazie klinicznym i różnej etiologii, charakteryzujących się występowaniem niekontrolowanych kurczów mięśni prowadzących do wymuszonego ułożenia części ciała. Istotną grupę wśród dystonii stanowią postacie genetycznie uwarunkowane. Do tej pory z występowaniem dystonii powiązano 25 loci chromosomowych i zidentyfikowano 16 genów, których mutacje są odpowiedzialne za jej różne postacie. Zidentyfikowane geny kodują m.in. białka związane ze szlakiem syntezy dopaminy, przewodnictwem synaptycznym, funkcjonowaniem cytoszkieletu, detoksykacją czy metabolizmem energetycznym komórki, co tłumaczyć może tak zróżnicowany patomechanizm i obraz kliniczny dystonii.
Wprowadzenie
Dystonie to termin określający nieprawidłowe, niezależne od woli, powtarzające się niekontrolowane ruchy, występujące w różnych częściach ciała, prowadzące do wymuszonego ich ułożenia. Termin ten oznacza także bardzo heterogenną grupę chorób neurologicznych związanych z zaburzeniami funkcjonowania jąder podstawy, co skutkuje dominacją ruchów dystonicznych w ich obrazie klinicznym.
Dystonie najczęściej występują sporadycznie, opisano jednak wiele rzadkich postaci rodzinnych o mendlowskim sposobie dziedziczenia, a dotąd zidentyfikowano 16 genów zaangażowanych w molekularne podłoże tej choroby.1 Pojęcie dystonii wprowadził Herman Oppenheim, który w 1911 r. opisał dystonię mięśniową deformacyjną (obecnie dystonia torsyjna typu 1), chociaż kluczowe opisy dla zrozumienia systematyki oraz rzucające światło na patogenezę przyniosły badania Sterlinga i Flauta.2,3 Podłoże molekularne, a zarazem pierwszy gen, którego mutacje powiązano z dystonią, zidentyfikowano jednak dopiero w roku 1994.4 Genetycznie uwarunkowane dystonie są chorobami rzadkimi, co w znacznym stopniu utrudnia badania. Brakuje dostatecznie licznych grup pacjentów jednorodnych pod względem klinicznym, co sprawia, że wykorzystanie i skuteczność klasycznych metod badawczych, takich jak analiza sprzężeń czy badania asocjacji, są ograniczone. W ostatnich latach rozwój technik sekwencjonowania DNA przyczynił się do znacznego postępu w poznaniu patologii molekularnej dystonii.
Celem prezentowanej pracy jest omówienie i charakterystyka podłoża molekularnego oraz patologii różnych postaci genetycznie uwarunkowanych dystonii.
Klasyfikacja dystonii
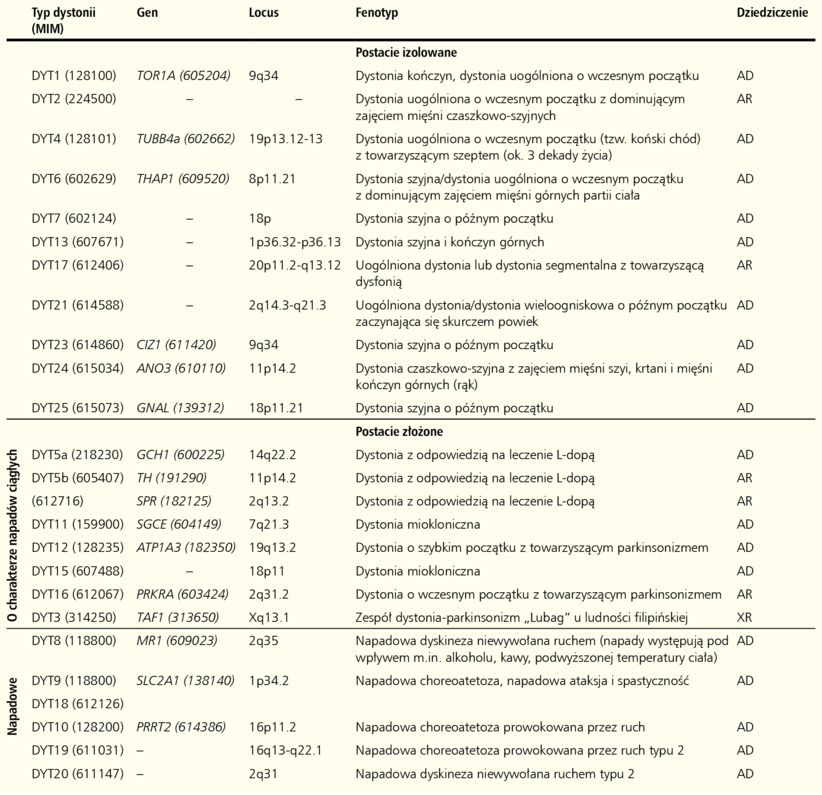
Tabela. Klasyfikacja molekularna dystonii uwarunkowanych genetycznie
Klasyfikację dystonii przez wiele lat opierano, zgodnie z zaleceniami EFNS (European Federation of Neurological Societies), na trzech kryteriach: etiologii, wieku zachorowania oraz anatomicznym zakresie występowania objawów.5 W 2013 roku dokonano aktualizacji tej klasyfikacji, opierając ją na dwóch podstawowych kryteriach diagnostycznych, tj. etiologii choroby oraz klinicznej charakterystyce objawów uwzględniającej: wiek wystąpienia objawów, anatomiczny zakres ich występowania, wzorzec czasowy rozwoju choroby oraz dodatkowe cechy, takie jak towarzyszące objawy lub zespoły neurologiczne.6 Wyróżniono postacie izolowane (obejmujące wcześniej dystonie pierwotne) i złożone (według poprzedniej nomenklatury określane jako dystonie-plus i dystonie napadowe) (tabela). Z punktu widzenia klinicysty istotny jest również podział dystonii na ogniskowe, segmentalne, wieloogniskowe, uogólnione, połowicze.
Postacie izolowane dystonii
Sporadyczne postacie izolowane dystonii stanowią grupę różnych chorób, w których dystonia jest jedynym objawem, a podłoże nie jest wynikiem procesu neurodegeneracyjnego. Do tej rodziny zaliczamy sześć genetycznie uwarunkowanych dystonii – dystonia torsyjna typ 1 (DYT1) oraz dystonie typu 4, 6, 23, 24 i 25.
Dystonia torsyjna typu 1
Dystonia torsyjna typu 1 (DYT1) jest dominującą postacią dystonii okresu rozwojowego. Pierwsze jej objawy mogą pojawić się w okresie dojrzewania, wieku młodzieńczym, a w sporadycznych przypadkach u dorosłych, zarówno pod postacią dystonii uogólnionej, jak i dystonii ogniskowej/segmentalnej, początkowo dotyczącej kończyny górnej albo dolnej, z czasem przyjmujące postać uogólnioną.7 Obraz kliniczny choroby może zmieniać się w czasie w zależności od wieku pacjenta, manifestacji objawów i progresji choroby. Choroba dziedziczy się w sposób autosomalny dominujący (AD) z penetracją wynoszącą 30-40%,8 a jej podłoże molekularne stanowią mutacje genu TOR1A. Gen ten koduje białko torsynę A (TOR1A), zlokalizowane w zakończeniach dendrytycznych i aksonalnych neuronów, gdzie prawdopodobnie bierze udział w zachowaniu integralności cytoszkieletu, transporcie aksonalnym i recyklingu pęcherzyków synaptycznych.9-11 Do tej pory opisano jedną patogenną mutację genu TOR1A w pozycji c.934-936GAGdel, skutkującą delecją jednego aminokwasu – kwasu glutaminowego (ΔE302/303) w białku i zaburzającą proces tworzenia funkcjonalnych oligomerów torsyny A.12 Patogenny charakter kilku innych mutacji zidentyfikowanych w tym genie pozostaje nadal niewyjaśniony.13-16
Dystonia typu 4
Dystonia typu 4 (DYT4) charakteryzuje się późniejszym wiekiem zachorowania (około trzeciej dekady życia), a jej głównym objawem jest dystonia uogólniona z współwystępującymi zaburzeniami chodu (tzw. koński chód) oraz dysfonią.17 Choroba dziedziczona jest w sposób autosomalny dominujący, a jej podłoże stanowią mutacje genu TUBB4 kodującego tubulinę 4β, podjednostkę składową mikrotubul.18 Zmutowane białko może powodować destabilizację cytoszkieletu przez niekorzystny wpływ na proces tworzenia i strukturę mikrotubul.19 TUBB4 ulega silnej ekspresji w mózgu, szczególnie zaś w móżdżku, którego dysfunkcja wiązana jest z występowaniem objawów dystonii.18
Dystonia typu 6
Dystonia typu 6 (DYT6) może ujawnić się między 4 a 55 rokiem życia. Ruchy dystoniczne w tej postaci choroby dotyczą najczęściej mięśni unerwianych przez nerwy czaszkowe, mięśnie szyi, krtani oraz kończyn górnych i mają tendencję do rozszerzania się na inne części ciała. U pacjentów często występuje dysfonia. W większości przypadków choroba dziedziczy się w sposób autosomalny dominujący z penetracją na poziomie około 60%.20 Podłoże molekularne stanowią w tym przypadku mutacje w genie THAP1. Białko kodowane przez ten gen wiąże się z DNA i bierze udział w regulacji transkrypcji własnego genu i wielu innych. Jednym z możliwych patomechanizmów DYT6 jest zaburzenie regulacji genów ulegających ekspresji w mózgu.21
Dystonia typu 23
Dystonia typu 23 (DYT23) ma postać dystonii ogniskowej z zajęciem mięśni szyi, której towarzyszyć może lekkie drżenie. Choroba ujawnia się we wczesnym dzieciństwie i w wieku dojrzałym, dziedziczona jest autosomalnie dominująco. Podłoże molekularne choroby najprawdopodobniej stanowią mutacje w genie CIZ1.22 Mutacje w tym genie zostały powiązane z występowaniem dystonii przez zastosowanie połączonych metod analizy sprzężeń i sekwencjonowania następnej generacji (next generation sequensing, NGS).23 Dla jednoznacznego potwierdzenia udziału mutacji genu CIZ1 w patogenezie dystonii wymagane są jednak badania na większej grupy pacjentów. Gen CIZ1 koduje białko zaangażowane w syntezę DNA i kontrolę cyklu komórkowego. Silną jego ekspresję zaobserwowano w neuronach ruchowych i móżdżku.22