



Co znajdziesz w artykule?
- Definicja, epidemiologia i etiologia nieimmunologicznego obrzęku płodu
- Przegląd klasycznych metod diagnostycznych
- W poszukiwaniu genetycznych przyczyn obrzęku – zastosowanie nowoczesnych metod diagnostyki molekularnej
Spis treści
Obrzęk płodu (łac. hydrops fetalis) definiowany jest jako gromadzenie się płynu w co najmniej dwóch jamach ciała, takich jak: jama otrzewnej, jama opłucnej, jama osierdzia, lub w jednej jamie ciała i tkance podskórnej. Nadmierna akumulacja płynu może być następstwem zwiększonego ciśnienia żylnego (np. w przypadku wad serca powodujących niewydolność krążenia), zmniejszonego ciśnienia onkotycznego osocza (np. w związku z niedostateczną produkcją białek osocza w przypadku zapalenia wątroby u
płodu z infekcją), jak również niedostatecznego odpływu chłonki (np. u płodów z torbielami limfatycznymi w przebiegu aberracji chromosomowych). Obrzęk może zarówno towarzyszyć mnogim wadom płodu, jak i występować u płodów o całkowicie prawidłowej budowie anatomicznej. Złożona etiologia obrzęku płodu obejmuje czynniki genetyczne, infekcyjne, kardiologiczne, metaboliczne i hematologiczne 1 .
Klasycznie obrzęk płodu ze względu na jego etiologię klasyfikowany jest jako obrzęk immunologiczny (IHF – immune hydrops fetalis), czyli związany z występowaniem u matki przeciwciał przeciwko krwinkom czerwonym płodu, lub jako obrzęk nieimmunologiczny (NIHF – non-immune hydrops fetalis), który obecnie występuje zdecydowanie częściej (w krajach rozwiniętych stanowi 85-90% przypadków obrzęku płodu) i na tym zagadnieniu koncentruje się niniejsze opracowanie. Brak ustalenia przyczyny NIHF może wpływać negatywnie na podejmowanie przyszłych decyzji prokreacyjnych. Dlatego tak ważne jest propagowanie wiedzy dotyczącej tej patologii, jak również objęcie pacjentek opieką specjalistyczną przez ośrodki dysponujące rozbudowanym zapleczem diagnostyczno-terapeutycznym.
Nieimmunologiczny obrzęk płodu występuje z częstością 1 : 1500-1 : 4000 urodzeń. W niektórych przypadkach, szczególnie jeśli w badaniach prenatalnych nie stwierdzono innych nieprawidłowości, obrzęk może ustąpić samoistnie i istnieje szansa na prawidłowy rozwój dziecka, jednak w większości przypadków NIHF wiąże się z wysoką, dochodzącą do 90%, śmiertelnością perinatalną 1, 2 . Rzadkim, lecz bardzo niebezpiecznym, powikłaniem NIHF jest tzw. zespół lustrzany u matki (mirror syndrome, inaczej zespół Ballantyne’a), w którym u ciężarnej rozwijają się objawy przypominające te obserwowane u płodu 3, 4 . Zespół lustrzany charakteryzuje się triadą objawów: obrzękiem płodu, obrzękiem łożyska oraz obrzękami u matki. Patogeneza tego zespołu nie jest w pełni poznana. Objawy kliniczne u matki mogą obejmować nadciśnienie tętnicze, białkomocz oraz uogólnione obrzęki, co może przypominać stan przedrzucawkowy. Objawem różnicującym jest obniżenie hematokrytu typowe dla zespołu lustrzanego w odróżnieniu od zagęszczenia krwi obserwowanego w stanie przedrzucawkowym. Wystąpienie zespołu lustrzanego stanowi poważne zagrożenie zarówno dla matki, jak i dla płodu, zwiększa śmiertelność płodów oraz zachorowalność matek 4 .
Medycyna prenatalna dysponuje obecnie szeroką gamą zaawansowanych interwencji wewnątrzmacicznych, które w wybranych przypadkach ciężkiego NIHF mogą poprawić rokowanie płodu oraz złagodzić objawy zespołu lustrzanego u ciężarnej lub im zapobiec 5 . Do kluczowych procedur należą transfuzje dopłodowe stosowane w przypadkach ciężkiej niedokrwistości płodu oraz shunty zakładane do jamy opłucnej w przypadku znacznego nagromadzenia płynu (hydrothorax). W niewielkiej części przypadków (np. NIHF w wyniku niedokrwistości o przejściowej przyczynie infekcyjnej, np. coraz częściej odnotowywane zakażenie parwowirusem B19) zastosowana metoda może prowadzić do pełnego wyleczenia płodu. Terapią o niekwestionowanej skuteczności, udowodnionej w licznych badaniach randomizowanych, jest również laserowa koagulacja połączeń naczyniowych w przypadku zespołu przetoczenia między płodami w ciąży bliźniaczej jednokosmówkowej. Należy jednak zaznaczyć, że możliwość zastosowania efektywnej terapii wewnątrzmacicznej dotyczy jedynie niewielkiego odsetka płodów z NIHF 5 .
Wybór odpowiedniej metody terapeutycznej zależy od precyzyjnego ustalenia etiologii NIHF oraz oceny stanu płodu i matki. Przed podjęciem decyzji o interwencji konieczne jest wnikliwe rozważenie zasadności takiego leczenia, w tym wykluczenie letalnych przyczyn NIHF, takich jak ciężkie wady genetyczne. Mimo postępów w diagnostyce prenatalnej i postnatalnej etiologia NIHF pozostaje niewyjaśniona w mniej więcej 20% przypadków 1 . Rozwój zaawansowanych technologii genetyki molekularnej, w tym sekwencjonowania eksomowego (ES – exome sequencing), a nawet sekwencjonowania całego genomu (GS – genome sequencing), może stanowić klucz do lepszego zrozumienia mechanizmów odpowiedzialnych za NIHF, co w przyszłości może wpłynąć na bardziej precyzyjne planowanie terapii.
Klasyczne metody diagnostyczne
W diagnostyce różnicowej obrzęku konieczne jest w pierwszej kolejności wykluczenie konfliktu serologicznego, co w praktyce sprowadza się do sprawdzenia obecności w surowicy matki przeciwciał przeciwko krwinkom czerwonym płodu 6, 7 .
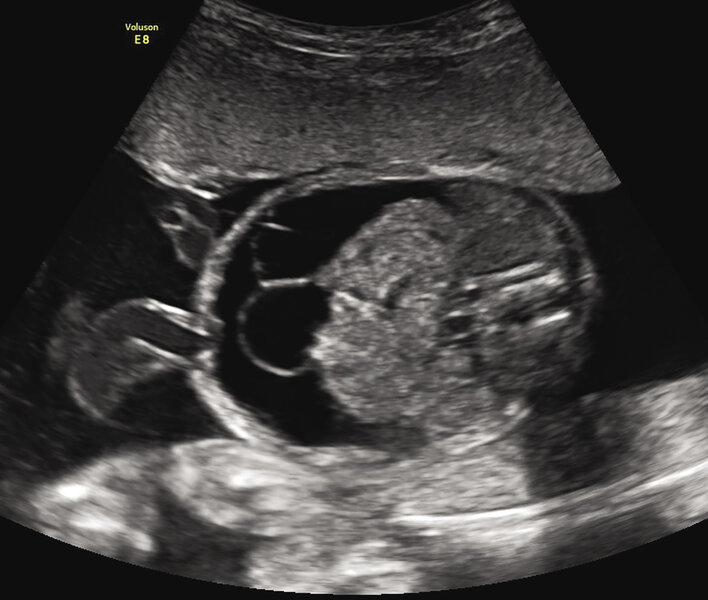
Rycina 1. 22 tydzień ciąży; C I, P I. Wodobrzusze u płodu spowodowane ciężką niedokrwistością w przebiegu konfliktu serologicznego w układzie Kell (Hb 1,7 mg/dl, Ht 5,5%, E 0,4 mln/μl). Pacjentka zakwalifikowana do leczenia wewnątrzmacicznego transfuzjami dopłodowymi – obrzęk ustąpił po trzeciej transfuzji
Niedokrwistość płodu, która jest jedną z podstawowych przyczyn obrzęku, może być spowodowana wieloma innymi czynnikami, takimi jak np. infekcja, przetoczenie płodowo-matczyne czy hemoglobinopatie i inne wrodzone choroby hematologiczne 1, 9, 10 (ryc. 2). Przyczynami niedokrwistości u płodu mogą też być zespół przetoczenia krwi między płodami (TTTS – twin-to-twin transfusion syndrome),

Rycina 2A, B. 23 tydzień ciąży; C II, P II. Wodobrzusze u płodu z niedokrwistością w przebiegu infekcji parwowirusem B19 (Hb 8 mg/dl, PLT 123 000 G/l). U starszego dziecka stwierdzono rumień zakaźny 6 tygodni wcześniej. Uwagę zwraca grube łożysko. Pacjentka zakwalifikowana do leczenia wewnątrzmacicznego transfuzjami dopłodowymi – obrzęk ustąpił po drugiej transfuzji
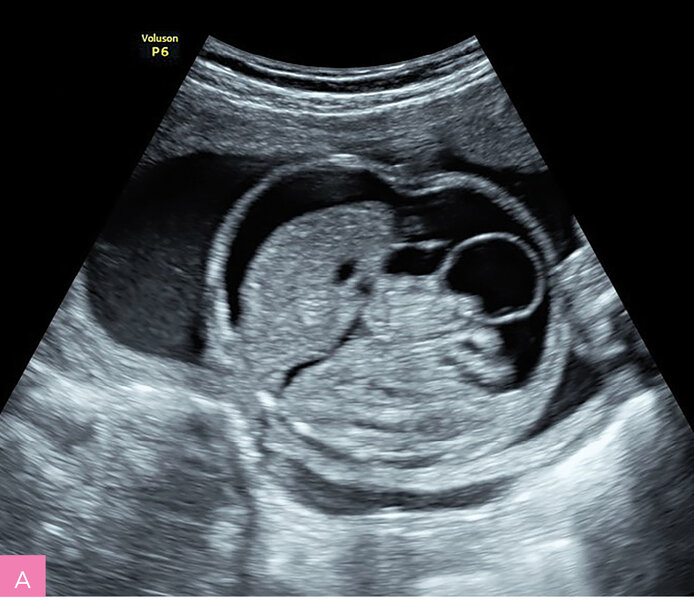
Rycina 2A, B. 23 tydzień ciąży; C II, P II. Wodobrzusze u płodu z niedokrwistością w przebiegu infekcji parwowirusem B19 (Hb 8 mg/dl, PLT 123 000 G/l). U starszego dziecka stwierdzono rumień zakaźny 6 tygodni wcześniej. Uwagę zwraca grube łożysko. Pacjentka zakwalifikowana do leczenia wewnątrzmacicznego transfuzjami dopłodowymi – obrzęk ustąpił po drugiej transfuzji
Podstawę diagnostyki NIHF stanowi szczegółowe badanie ultrasonograficzne, które pozwala na ocenę anatomii płodu oraz wykrycie ewentualnych towarzyszących wad strukturalnych. Istnieje wiele nieprawidłowości będących bezpośrednią przyczyną zwiększonej akumulacji płynu w jamach ciała i są to zazwyczaj stany prowadzące do niewydolności krążenia, takie jak zaburzenia rytmu serca (w szczególności tachyarytmie), niektóre wady serca (np. ciężka stenoza aortalna lub krytyczne zwężenie zastawki płucnej u płodu), a także wady związane z krążeniem hiperkinetycznym, w tym bogato unaczynione guzy płodu lub łożyska (np. lity potworniak kości krzyżowej). Malformacja żyły Galena (tętniak żyły Galena) to niezwykle rzadka patologia płodu charakteryzująca się występowaniem nieprawidłowego połączenia pomiędzy tętnicami a żyłą Galena – krótkim naczyniem utworzonym w miejscu spływu żył mózgu wewnętrznych i żył podstawnych Rosenthala, odprowadzającym krew żylną do zatoki prostej.
Rycina 3A, B. 30 tydzień ciąży; C I, P I. Pacjentka skierowana z powodu kardiomegalii i nieprawidłowego obrazu ośrodkowego układu nerwowego. W badaniu stwierdzono malformację tętniczo-żylną Galena (tętniak żyły Galena) i wtórną niewydolność krążenia (powiększona sylwetka serca, znacznego stopnia niedomykalność zastawki trójdzielnej i mitralnej, wsteczny przepływ w przewodzie żylnym). Pacjentkę poinformowano o poważnym rokowaniu (prawdopodobny zgon po urodzeniu). Poród odbył się w 37 tygodniu ciąży. Zgon noworodka nastąpił w pierwszej godzinie życia
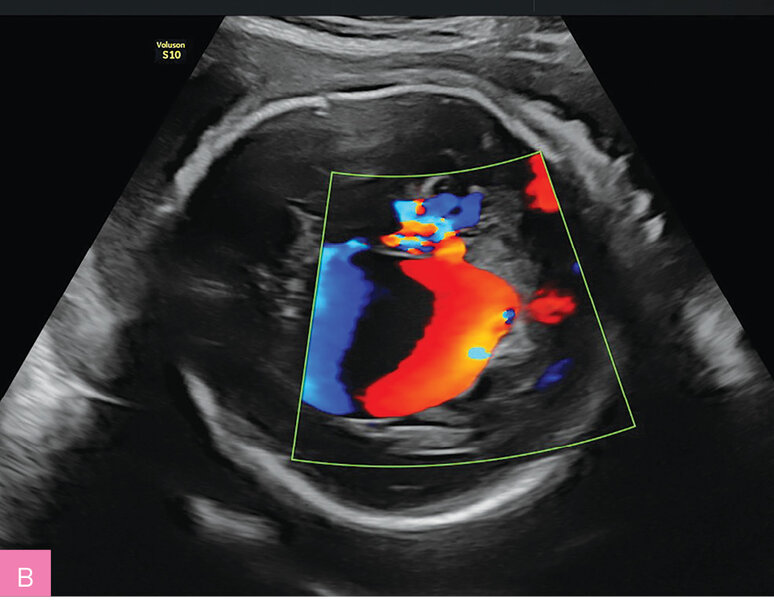
Rycina 3A, B. 30 tydzień ciąży; C I, P I. Pacjentka skierowana z powodu kardiomegalii i nieprawidłowego obrazu ośrodkowego układu nerwowego. W badaniu stwierdzono malformację tętniczo-żylną Galena (tętniak żyły Galena) i wtórną niewydolność krążenia (powiększona sylwetka serca, znacznego stopnia niedomykalność zastawki trójdzielnej i mitralnej, wsteczny przepływ w przewodzie żylnym). Pacjentkę poinformowano o poważnym rokowaniu (prawdopodobny zgon po urodzeniu). Poród odbył się w 37 tygodniu ciąży. Zgon noworodka nastąpił w pierwszej godzinie życia
Najczęstszą aberracją chromosomową w NIHF jest monosomia X (ryc. 4), ale obrzęk może występować również u płodów z innymi aberracjami,
Rycina 4. 22 tydzień ciąży; C III, P II. Ogromny wodniak karku u płodu z monosomią X (zespół Turnera). Obumarcie wewnątrzmaciczne nastąpiło w 22 tygodniu ciąży
Rycina 5. 15 tydzień ciąży; C II, P I. Uogólniony obrzęk płodu z trisomią 18 (zespół Edwardsa). Uwagę zwraca nieprawidłowy trójkątny kształt czaszki
Aberracje chromosomowe możliwe do wykrycia za pomocą aCGH są najczęstszą przyczyną obrzęku płodu w I trymestrze ciąży 1, 12 . W przypadku obrzęku rozwijającego się na późniejszym etapie ciąży diagnostyka może wymagać bardziej zaawansowanych narzędzi, takich jak sekwencjonowanie eksomowe (ES), a nawet sekwencjonowanie genomowe (GS), które zostaną omówione w dalszej części opracowania. NIHF może bowiem występować w różnych zespołach związanych z patogennymi wariantami w specyficznych genach, których wykrycie nie jest możliwe przy użyciu klasycznego badania cytogenetycznego lub aCGH (w tzw. chorobach monogenowych) 14 .
Znanych jest ponad 80 jednostek chorobowych dziedziczonych monogenowo, które mogą się wiązać z występowaniem NIHF, a do najczęściej opisywanych w literaturze należą tzw. RASopatie 11, 12 . Są to rzadkie, dziedziczne schorzenia spowodowane patogennymi wariantami w genach kodujących białka szlaku sygnalizacyjnego RAS/MAPK prowadzącymi do jego nadmiernej aktywacji. Szlak RAS/MAPK odgrywa kluczową rolę w regulacji proliferacji, różnicowania, dojrzewania i apoptozy komórek, a jego zaburzenia skutkują nieprawidłowym rozwojem tkanek i narządów, a także zwiększonym ryzykiem nowotworów. Jednym z najczęściej występujących zaburzeń w tej grupie jest zespół Noonan, którego cechami charakterystycznymi są: niski wzrost, dysmorfia twarzy, wady serca i opóźnienie rozwoju. W okresie prenatalnym u płodów z tym zespołem często obserwuje się zwiększoną przezierność karkową (NT – nuchal translucency) w I trymestrze ciąży, a w II trymestrze – wodniaka karku, obrzęk tkanki podskórnej oraz obecność hydrothorax. Diagnostyka genetyczna w kierunku zespołu Noonan obejmuje analizę takich genów, jak: PTPN11, SOS1, KRAS, RAF1, BRAF, MEK1/2 oraz NF1, choć patogenne warianty mogą występować również w innych genach 11, 15 .
Dysplazje kostne obejmują ponad 400 różnych jednostek chorobowych. Patogenne warianty genetyczne prowadzą do objawowych zaburzeń formowania układu szkieletowego, a zmienna penetracja i ekspresja poszczególnych wariantów odpowiada za zróżnicowane nasilenie objawów. Niektóre choroby kości diagnozowane są dopiero po urodzeniu, jednak część dysplazji, w tym dysplazja tanatoforyczna, dysplazja kampomeliczna czy osteogenesis imperfecta, charakteryzuje się ciężkim lub letalnym przebiegiem. Zespoły te są często związane z defektami strukturalnymi i hemodynamicznymi, które mogą przyczyniać się do obrzęku, szczególnie w połączeniu z wadami serca lub układu limfatycznego 16 . Mutacje w genach, takich jak FLT4, który jest związany z wrodzonymi wadami układu chłonnego, mogą prowadzić do uogólnionego obrzęku płodu wynikającego z niewydolności drenażu limfatycznego. Ponadto mutacje w genach odpowiedzialnych za rozwój układu sercowo-naczyniowego, takich jak NKX2-5 czy MYH7, mogą prowadzić do wad wrodzonych serca i wtórnego obrzęku płodu.
Choroby metaboliczne stanowią grupę rzadkich zaburzeń, w których patogenne warianty genetyczne prowadzą na poziomie komórkowym do dysfunkcji enzymatycznych lub zaburzeń w procesach metabolicznych. Do najczęściej opisywanych w piśmiennictwie należą: wrodzone defekty metabolizmu glikogenu, wrodzone zaburzenia glikozylacji białek (CDG – congenital disorders of glycosylation) oraz zaburzenia w metabolizmie lipidów i lizosomalne choroby spichrzeniowe, takie jak choroba Niemanna-Picka typu C czy choroba Gauchera. W tych przypadkach obrzęk płodu jest często skutkiem uogólnionej dysfunkcji narządowej prowadzącej do upośledzenia funkcji wątroby, serca lub układu limfatycznego 17 . Przed wprowadzeniem zaawansowanych metod genetyki molekularnej diagnostyka chorób metabolicznych opierała się na badaniu aktywności wybranych enzymów w płynie owodniowym. Ze względu na celowany charakter badania i konieczność pozyskania znacznej objętości płynu owodniowego metoda ta miała jednak ograniczone znaczenie w okresie prenatalnym. Obecnie prowadzone są badania nad zastosowaniem terapii enzymatycznej w leczeniu chorób lizosomalnych już w okresie prenatalnym. Dr Tippi MacKenzie i jej zespół z University of California w San Francisco prowadzą przełomowe badania w zakresie chorób metabolicznych, takich jak lizosomalne choroby spichrzeniowe. Badacze w szczególności koncentrują się na wewnątrzmacicznym podawaniu enzymów zastępczych (IUERT – in utero enzyme replacement therapy), co ma na celu zapobiegnięcie uszkodzeniom ośrodkowego układu nerwowego na etapie niedojrzałej bariery krew–mózg. Wyniki wstępnych badań klinicznych (NCT04532047) wskazują, że IUERT może być skutecznym narzędziem w prewencji nieodwracalnych uszkodzeń narządów i rozwoju odpowiedzi immunologicznej na białka terapeutyczne 18 .
Ustalenie genetycznego podłoża NIHF ma kluczowe znaczenie dla poradnictwa genetycznego, pozwala na lepsze oszacowanie ryzyka nawrotu oraz planowanie postępowania w przyszłych ciążach 11, 14, 19 . Choroby monogenowe dziedziczone autosomalnie recesywnie wiążą się z wysokim ryzykiem powtórzenia w kolejnej ciąży, które wynosi 25%. Ustalenie nosicielstwa chorób dziedziczonych autosomalnie recesywnie umożliwia diagnostykę preimplantacyjną w przypadku zastosowania metod wspomaganego rozrodu bądź celowaną diagnostykę prenatalną z materiału pozyskanego na drodze biopsji kosmówki, co zwiększa szanse na urodzenie zdrowego dziecka. Ze względu na możliwość mozaikowatości ograniczonej do gonad NIHF może pojawić się w kolejnej ciąży również w przypadku chorób dziedziczonych autosomalnie dominująco. Zidentyfikowanie podłoża genetycznego NIHF ma kluczowe znaczenie dla zrozumienia patomechanizmu obrzęku i może otworzyć drogę do specyficznych terapii enzymatycznych w okresie pre- i postnatalnym.
Nowoczesne metody diagnostyki molekularnej
Sekwencjonowanie eksomowe to zaawansowana technika genetyczna umożliwiająca analizę wszystkich sekwencji kodujących białka w celu identyfikacji wariantów o potencjalnym charakterze patogennym 20 . Metoda ta znajduje szczególne zastosowanie w diagnostyce chorób monogenowych, które nie są możliwe do wykrycia z pomocą klasycznych metod cytogenetycznych, takich jak prążkowa analiza kariotypu czy kariotyp molekularny z użyciem mikromacierzy (aCGH).
Od kilku lat ES zyskuje coraz większe znaczenie w diagnostyce prenatalnej NIHF. Częstość wykrywania patogennych wariantów genetycznych w różnych populacjach jest zróżnicowana i zależy od obecności towarzyszących wad strukturalnych 21, 22, 23, 24, 25, 26 . Przykładowo w badaniu Lord i wsp. odsetek zidentyfikowanych patogennych wariantów wynosił 9% 23 , podczas gdy w badaniu HYDROPS osiągnął nawet 50% 24 . W badaniu przeprowadzonym przez Deng i wsp. w grupie 109 płodów z NIHF w podgrupie 21 przypadków z prawidłowym wynikiem aCGH zidentyfikowano 3 patogenne warianty (14,3%) oraz 2 warianty o nieznanej patogenności (VUS – variants of uncertain significance), co stanowiło dodatkową wartość diagnostyczną 2 . Z kolei Sparks i wsp. w swoim prospektywnym badaniu wykazali, że zastosowanie ES pozwala na postawienie diagnozy genetycznej w 29% przypadków 25 .
Na podstawie metaanalizy Mone i wsp. potwierdzili, że ES przynosi dodatkową wartość diagnostyczną w 39% przypadków płodów z NIHF i towarzyszącymi wadami strukturalnymi 26 . Te dane jednoznacznie podkreślają znaczenie ES jako niezastąpionego narzędzia w kompleksowej ocenie etiologii NIHF. W innym badaniu, przeprowadzonym przez Liu i wsp., przeanalizowano 53 przypadki niewyjaśnionego NIHF, w tym 39 płodów spełniało ścisłą definicję NIHF, a 14 charakteryzowało się jedynie podwyższoną NT i/lub obecnością wodniaka karku. Patogenne warianty genetyczne zidentyfikowano w 23% przypadków (12 z 53), a potencjalnie patogenne w dodatkowych 13% (7 z 53). Co istotne, w 3 przypadkach (7%) diagnozę postawiono dopiero po ponownej analizie danych. Najwyższy odsetek wykrytych wariantów patogennych i potencjalnie patogennych (43%) odnotowano w grupie płodów z obrzękiem skóry oraz z podwyższoną NT lub wodniakiem karku. W tej grupie liczącej 14 płodów rozpoznanie ustalono w 6 przypadkach. Natomiast w klasycznych przypadkach NIHF częstość wykrycia patogennych wariantów wynosiła 15% (6 z 39). W badaniu tym wskazano również na wpływ zastosowania ES na podejmowane decyzje kliniczne, gdyż odsetek terminacji ciąży w grupie z wykrytymi patogennymi wariantami genetycznymi był znacząco wyższy niż w grupie z prawidłowym wynikiem ES (92% vs 54%), a odsetek żywych urodzeń w tych grupach wynosił odpowiednio 8% i 37% 27 .
W ramach badań własnych prowadzonych we współpracy z Zakładem Genetyki Medycznej Instytutu Matki i Dziecka w Warszawie przeanalizowano grupę 10 płodów z rozpoznanym NIHF oraz prawidłowym wynikiem aCGH. Średni wiek ciąży w momencie wykrycia obrzęku wynosił 18 tygodni. U 8 z 10 płodów (80%) zaobserwowano dodatkowe wady strukturalne, co wskazuje na częste współwystępowanie anomalii morfologicznych u płodów z NIHF. Najczęściej stwierdzano:
Rycina 6A, B. 33 tydzień ciąży; C I, P I. Obrzęk płodu od 20 tygodnia ciąży. Wielowodzie. Uwagę zwracało wyprostne ustawienie kończyn przez cały czas badania USG. Wynik badania aCGH prawidłowy. Ze względu na podejrzenie choroby skórno-mięśniowej o podłożu monogenowym wykonano ES – stwierdzono patogenny wariant w genie ACTA1 powodujący letalną miopatię nemalinową. Poród przedwczesny w 33 tygodniu ciąży, okołoporodowy zgon płodu
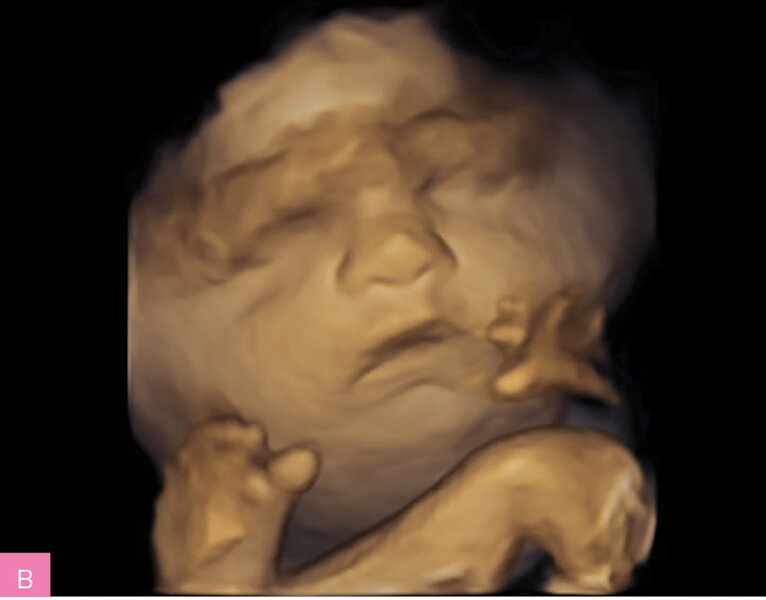
Rycina 6A, B. 33 tydzień ciąży; C I, P I. Obrzęk płodu od 20 tygodnia ciąży. Wielowodzie. Uwagę zwracało wyprostne ustawienie kończyn przez cały czas badania USG. Wynik badania aCGH prawidłowy. Ze względu na podejrzenie choroby skórno-mięśniowej o podłożu monogenowym wykonano ES – stwierdzono patogenny wariant w genie ACTA1 powodujący letalną miopatię nemalinową. Poród przedwczesny w 33 tygodniu ciąży, okołoporodowy zgon płodu
Istnieje kilka wyzwań związanych z interpretacją wyników ES 19, 20 . Choroby o dziedziczeniu jednogenowym mogą charakteryzować się zmienną ekspresją i penetracją, co oznacza, że ta sama mutacja genetyczna może prowadzić do różnych fenotypów klinicznych. Dodatkowo baza danych genetycznych jest ciągle uaktualniana, w związku z czym warianty genetyczne, które obecnie są klasyfikowane jako te o nieznanej patogenności (VOUS – variants of unknown significance), w przyszłości mogą zostać uznane za patogenne na podstawie nowych doniesień naukowych. Ograniczona liczba badań dotyczących prenatalnego wykorzystania ES utrudnia pełne zrozumienie związku między określonymi wariantami genetycznymi a fenotypem NIHF. Rozwój genetyki molekularnej i coraz większa dostępność baz danych wariantów genetycznych pozwalają na bardziej precyzyjną diagnostykę, która z czasem stanie się rutynową częścią diagnostyki prenatalnej 19 .
Wprowadzenie ES do diagnostyki NIHF znacząco zwiększyło wykrywalność podłoża genetycznego. ES obejmuje jednak jedynie sekwencje kodujące białka, które stanowią zaledwie 1,5-2% całego genomu. Z tego względu diagnostyka oparta wyłącznie na ES może pomijać potencjalnie istotne zmiany genetyczne zlokalizowane w sekwencjach niekodujących.
Sekwencjonowanie genomowe umożliwia szczegółową analizę całego genomu, w tym sekwencji niekodujących. Metoda ta pozwala na wykrycie nie tylko mutacji punktowych, lecz także aberracji strukturalnych, takich jak: insercje, delecje czy zrównoważone translokacje, które mogą być trudne do zidentyfikowania za pomocą aCGH. Ponadto GS pozwala na detekcję sekwencji powtórzonych, które potencjalnie mogą leżeć u podstaw NIHF, a których analiza jest poza zasięgiem ES czy aCGH.
Dotychczas zastosowanie GS w diagnostyce NIHF zostało opisane w jednym opublikowanym badaniu. Westenius i wsp. oceniali przydatność GS w grupie 23 płodów z NIHF, u których wcześniejsze badania aCGH nie wykazały nieprawidłowości. GS umożliwiło wykrycie nieprawidłowych wariantów genetycznych w 12 przypadkach (53%) 29 . Warto jednak zauważyć, że analiza w tym badaniu była ograniczona do panelu 281 genów związanych z NIHF, co sugeruje, że pełne wykorzystanie potencjału GS mogłoby dostarczyć jeszcze bardziej szczegółowych danych diagnostycznych. Podczas badań własnych finansowanych z Narodowego Centrum Nauki w ramach grantu naukowego MINIATURA 7 w Klinice Ginekologii i Ginekologii Onkologicznej Centrum Medycznego Kształcenia Podyplomowego w Warszawie przeprowadzono GS u 14 płodów z NIHF o niewyjaśnionej przyczynie.
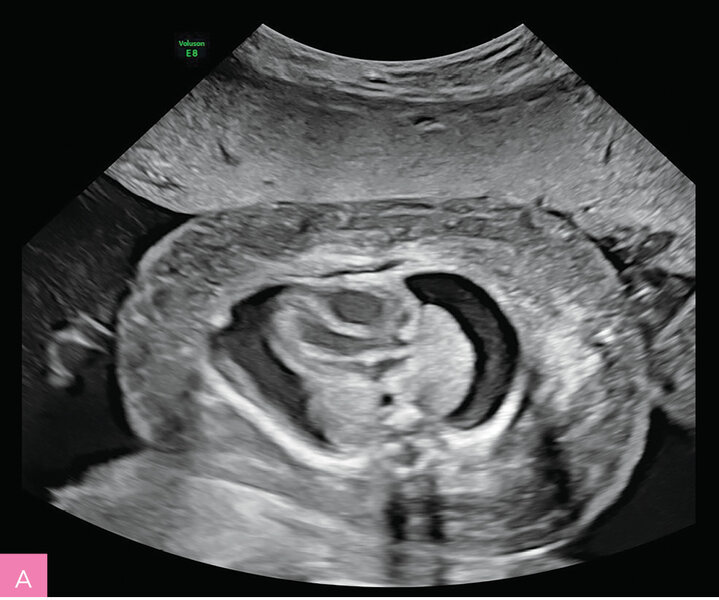
Rycina 7A, B. 23 tydzień ciąży (A) i 32 tydzień ciąży (B); C II, P II. Obrzęk płodu obserwowany od 20 tygodnia ciąży. Łagodna niedokrwistość płodu. Nie stwierdzono wad strukturalnych. Uwagę zwracały ogromny obrzęk tkanki podskórnej, obejmujący twarz, kończyny górne i dolne oraz mosznę, a także znaczne powiększenie wątroby. Od 30 tygodnia ciąży obserwowano wielowodzie. Wynik badania aCGH prawidłowy. Ujemne wyniki badania w kierunku wybranych chorób metabolicznych z płynu owodniowego i badania przesiewowego w kierunku najczęstszych chorób monogenowych z kropli krwi płodu, pobranej na drodze kordocentezy. PPROM w 35 tygodniu ciąży. Zgon okołoporodowy. Za pomocą GS udało się ustalić znany patogenny intronowy wariant w genie PIEZO1 odpowiadający za uogólnioną dysplazję limfatyczną, co odpowiadało obserwowanemu fenotypowi
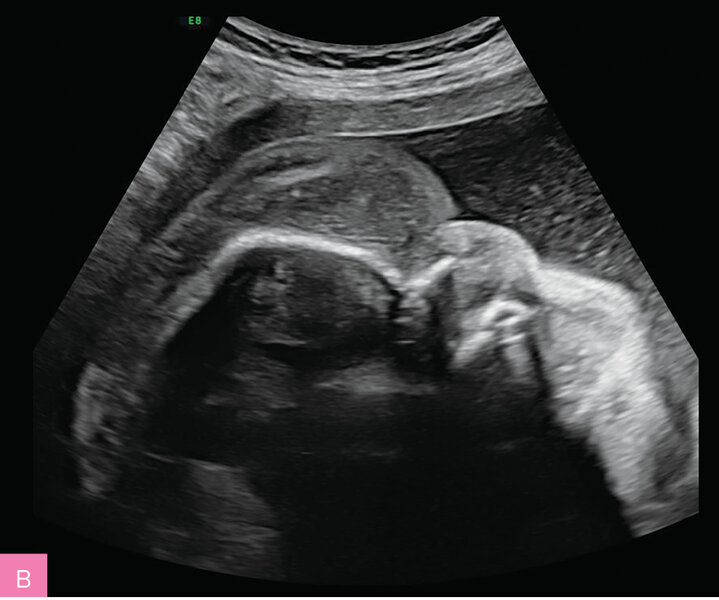
Rycina 7A, B. 23 tydzień ciąży (A) i 32 tydzień ciąży (B); C II, P II. Obrzęk płodu obserwowany od 20 tygodnia ciąży. Łagodna niedokrwistość płodu. Nie stwierdzono wad strukturalnych. Uwagę zwracały ogromny obrzęk tkanki podskórnej, obejmujący twarz, kończyny górne i dolne oraz mosznę, a także znaczne powiększenie wątroby. Od 30 tygodnia ciąży obserwowano wielowodzie. Wynik badania aCGH prawidłowy. Ujemne wyniki badania w kierunku wybranych chorób metabolicznych z płynu owodniowego i badania przesiewowego w kierunku najczęstszych chorób monogenowych z kropli krwi płodu, pobranej na drodze kordocentezy. PPROM w 35 tygodniu ciąży. Zgon okołoporodowy. Za pomocą GS udało się ustalić znany patogenny intronowy wariant w genie PIEZO1 odpowiadający za uogólnioną dysplazję limfatyczną, co odpowiadało obserwowanemu fenotypowi
Podsumowanie
Nieimmunologiczny obrzęk płodu nadal stanowi istotne wyzwanie diagnostyczne. Prenatalne ustalenie przyczyny obrzęku umożliwia zaplanowanie optymalnego postępowania i ewentualne wdrożenie leczenia, a także pozwala na oszacowanie ryzyka powtórzenia się NIHF w kolejnych ciążach. Diagnostyka genetyczna, zwłaszcza ES, otwiera nowe możliwości w identyfikacji patogennych wariantów genetycznych, które mogą być przyczyną obrzęku. Wprowadzenie ES do diagnostyki prenatalnej pomaga w ustaleniu rozpoznania w przypadkach, w których inne metody nie przyniosły rezultatu, szczególnie u płodów z towarzyszącymi wadami strukturalnymi. Ponowna analiza danych w świetle aktualnej wiedzy może dodatkowo zwiększyć skuteczność diagnostyczną. Dalszy rozwój badań genetycznych, w tym GS, może się przyczynić do znacznego postępu w opiece nad pacjentkami z rozpoznanym NIHF.
Abstract
Diagnostic challenges in non-immune hydrops fetalis with special regard to advanced genetic diagnostics
Hydrops fetalis is defined as the accumulation of fluid in at least two body cavities, such as the peritoneal cavity, pleural cavity or pericardial cavity, or in one body cavity and subcutaneous tissue. Foetal oedema can accompany multiple foetal defects or be detected in an otherwise healthy foetus. This article focuses on diagnostic and therapeutic possibilities in non–immune hydrops fetalis (NIHF), which accounts for 85-90% of cases of foetal oedema in developed countries. NIHF has a complex aetiology involvjng genetic, infectious, cardiac, hematological and metabolic causes. Despite diagnostic progress, the cause of NIHF cannot be ascertained in 20% of cases.
This article stresses the importance of prenatal diagnostics based on modern molecular genetic techniques, such as exome sequencing (ES) and whole-genome sequencing (GS). These techniques allow for the identification of genetic variants in monogenic diseases, such as RAS-opathies (e.g., Noonan syndrome) or bone dysplasias, that may be associated with hyodrops fetalis. The advent of ES has significantly improved diagnostic accuracy, especially in the presence of concomitant structural defects.
Additionally, we discuss a rare but very dangerous complication of NIHF in the form of so-called mirror syndrome (Ballantyne syndrome) in the expectant mother, who develops symptoms resembling those in the foetus. The onset of mirror syndrome represents a serious threat to both the mother and the foetus, increasing both foetal mortality and maternal morbidity.
In summary, advanced genetic techniques open new possibilities in the diagnosis and treatment of NIHF, which is of significant importance for the foetal prognosis and the planning of future pregnancies. Further progress in techniques such as GS can contribute to a better understanding of the underlying pathomechanisms and the development of more effective pre- and postnatal therapies.
- 1. Bellini C, Donarini G, Paladini D, et al. Etiology of non-immune hydrops fetalis: an update. Am J Med Genet A 2015;167A(5):1082-8. doi: 10.1002/ajmg.a.36988
- 2. Deng Q, Fu F, Yu Q, et al. Nonimmune hydrops fetalis: genetic analysis and clinical outcome. Prenat Diagn 2020;40(7):803-12. doi: 10.1002/pd.5691
- 3. Chen R, Liu M, Yan J, et al. Clinical characteristics of mirror syndrome: a retrospective study of 16 cases. J Obstet Gynaecol 2021;41(1):73-6. doi: 10.1080/01443615.2020.1718621
- 4. Hirata G, Aoki S, Sakamaki K, et al. Clinical characteristics of mirror syndrome: a comparison of 10 cases of mirror syndrome with non-mirror syndrome fetal hydrops cases. J Matern Fetal Neonatal Med 2016;29(16):2630-4. doi: 10.3109/14767058.2015.1095880
- 5. Sichitiu J, Alkazaleh F, de Heus R, et al. Maternal "mirror" syndrome: evaluating the benefits of fetal therapy. Prenat Diagn 2024;44(8):979-87. doi: 10.1002/pd.6589
- 6. Adani SN, Mohd Ashari NS, Johan MF, et al. Red blood cell alloimmunization in pregnancy: a review of the pathophysiology, prevalence and risk factors. Cureus 2024 ;16(5):e60158. doi: 10.7759/cureus.60158
- 7. Moise KJ Jr, Argoti PS. Management and prevention of red cell alloimmunization in pregnancy: a systematic review. Obstet Gynecol 2012;120(5):1132-9. doi: 10.1097/aog.0b013e31826d7dc1
- 8. Vlachodimitropoulou E, Shehata N, Ryan G, et al. Management of pregnancies with anti-K alloantibodies and the predictive value of anti-K titration testing. Lancet Haematol 2024;11(11):e873-7. doi: 10.1016/S2352-3026(24)00239-4
- 9. Kagan KO, Hoopmann M, Geipel A, et al. Prenatal parvovirus B19 infection. Arch Gynecol Obstet 2024;310(5):2363-71. doi: 10.1007/s00404-024-07644-6
- 10. Bijok J, Warakomska M, Massalska D, et al. Foeto-maternal haemorrhage: an unexpected challenge. J Obstet Gynaecol 2017;37(6):818-20. doi: 10.1080/01443615.2017.1289159
- 11. Chen CP. Chromosomal abnormalities associated with fetal pleural effusion (I): general overview. Taiwan J Obstet Gynecol 2024;63(2):165-7. doi: 10.1016/j.tjog.2024.01.009
- 12. Chen CP. Chromosomal abnormalities associated with fetal pleural effusion (II): specific and non-specific chromosome aberrations. Taiwan J Obstet Gynecol 2024;63(2):168-73. doi: 10.1016/j.tjog.2024.01.010
- 13. Massalska D, Bijok J, Ilnicka A, et al. Triploidy – variability of sonographic phenotypes. Prenat Diagn 2017;37(8):774-80. doi: 10.1002/pd.5080
- 14. Chen CP. Syndromic and single gene disorders associated with fetal pleural effusion (I): Noonan syndrome, RASopathy and congenital lymphatic anomalies. Taiwan J Obstet Gynecol 2024;63(2):174-7. doi: 10.1016/j.tjog.2024.01.011
- 15. Pevec U, Rozman N, Gorsek B, et al. RASopathies: presentation at the genome, interactome and phenome levels. Mol Syndromol 2016;7(2):72-9. doi: 10.1159/000445733
- 16. Kucińska-Chahwan A, Roszkowski T, Nowakowska B, et al. Genetic causes of the skeletal system abnormalities diagnosed by prenatal sonography with the use of exome sequencing: single institution experience. Ultrasound Obstet Gynecol 2021. doi: 10.1002/uog.23722
- 17. Epstein Weiss T, Erez O, Hazan I, et al. Characterization of pregnancy outcome of women with an offspring with inborn errors of metabolism: a population-based study. Front Genet 2022;13:1030361. doi: 10.3389/fgene.2022.1030361
- 18. Herzeg A, Borges B, Lianoglou BR, et al. Intrauterine enzyme replacement therapies for lysosomal storage disorders: current developments and promising future prospects. Prenat Diagn 2023;43(13):1638-49. doi: 10.1002/pd.6460
- 19. Chong JX, Buckingham KJ, Jhangiani SN, et al. The genetic basis of mendelian phenotypes: discoveries, challenges and opportunities. Am J Hum Genet 2015;97(2):199‐215. doi: 10.1016/j.ajhg.2015.06.009
- 20. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17(5):405‐24. doi: 10.1038/gim.2015.30
- 21. Petrovski S, Aggarwal V, Giordano JL, et al. Whole-exome sequencing in the evaluation of fetal structural anomalies: a prospective cohort study. Lancet 2019;393(10173):758-67. doi: 10.1016/S0140-6736(18)32042-7
- 22. Best S, Wou K, Vora N, et al. Promises, pitfalls and practicalities of prenatal whole exome sequencing. Prenat Diagn 2018;38(1):10-9. doi: 10.1002/pd.5102
- 23. Lord J, McMullan DJ, Eberhardt RY, et al. Prenatal exome sequencing analysis in fetal structural anomalies detected by ultrasonography (PAGE): a cohort study. Lancet 2019;393(10173):747‐57. doi: 10.1016/S0140-6736(18)31940-8
- 24. Al-Kouatly HB, Makhamreh MM, Rice SM, et al. High diagnosis rate for nonimmune hydrops fetalis with prenatal clinical exome from the Hydrops-Yielding Diagnostic Results of Prenatal Sequencing (HYDROPS) study. Genet Med 2021;23(7):1325-33. doi: 10.1038/s41436-021-01121-0
- 25. Sparks TN, Lianoglou BR, Adami RR, et al.; University of California Fetal–Maternal Consortium; University of California, San Francisco Center for Maternal–Fetal Precision Medicine. Exome sequencing for prenatal diagnosis in nonimmune hydrops fetalis. N Engl J Med 2020;383(18):1746-56. doi: 10.1056/NEJMoa2023643
- 26. Mone F, Eberhardt RY, Hurles ME, et al. Fetal hydrops and the Incremental yield of Next-generation sequencing over standard prenatal Diagnostic testing (FIND) study: prospective cohort study and meta-analysis. Ultrasound Obstet Gynecol 2021;58(4):509-18. doi: 10.1002/uog.23652
- 27. Liu C, Huang Y, Wang Y, et al. Exome sequencing for nonimmune hydrops fetalis and clinical utility of data reanalysis. QJM 2024:hcae187. doi: 10.1093/qjmed/hcae187
- 28. Kucińska-Chahwan A, Geremek M, Roszkowski T, et al. Implementation of exome sequencing in prenatal diagnosis and impact on genetic counseling: the Polish experience. Genes 2022;13(5):724. doi: 10.3390/genes13050724
- 29. Westenius E, Sahlin E, Conner P, et al. Diagnostic yield using whole-genome sequencing and in-silico panel of 281 genes associated with non-immune hydrops fetalis in clinical setting. Ultrasound Obstet Gynecol 2022;60(4):487-93. doi: 10.1002/uog.24911
Następny artykuł:

 Dodaj do ulubionych
Dodaj do ulubionych
