



Co znajdziesz w artykule?
- Wielu specjalistów zajmujących się opieką nad pacjentami ze stwardnieniem zanikowym bocznym nie jest świadomych współistnienia zaburzeń poznawczych i zachowania z objawami ruchowymi, co wpływa na brak zrozumienia zgłaszanych przez chorych i/lub opiekunów dolegliwości
- Badania nad zaburzeniami funkcji poznawczych i zachowania, ich profilem i częstością występowania otępienia, w tym zwłaszcza otępienia czołowo-skroniowego w stwardnieniu zanikowym bocznym, mają ogromne znaczenie w zrozumieniu patomechanizmu tych schorzeń i dają nadzieję na znalezienie skutecznych metod ich leczenia
Spis treści
- Obraz kliniczny i profile zaburzeń ruchowych w chorobie neuronu ruchowego
- Zaburzenia poznawcze i językowe w SLA
- Zaburzenia zachowania w SLA
- Znaczenie oceny funkcji poznawczych w SLA
- Kryteria diagnostyczne zespołów klinicznych ze spektrum SLA i otępienia czołowo-skroniowego
- Badanie neuropsychologiczne pacjenta z SLA
- Ocena zaburzeń zachowania w SLA
- Znaczenie badania genetycznego w diagnostyce różnicowej SLA-FTSD
- Znaczenie badań obrazowych w diagnostyce SLA-FTSD
- Znaczenie kliniczne zaburzeń poznawczych w SLA
- Podsumowanie
Przez wiele dziesięcioleci uznawano, że pacjenci z chorobami neuronu ruchowego zachowują pełną sprawność poznawczą przez cały czas trwania choroby. Na podstawie wyników badań z ostatnich 20-30 lat wykazano, że chorzy z rozpoznaniem stwardnienia zanikowego bocznego (SLA – sclerosis lateralis amyotrophica; ALS – amyotrophic lateral sclerosis) mogą ujawniać deficyty poznawcze, językowe i zaburzenia zachowania, które przyczyniają się do ich niesprawności. Te dysfunkcje mogą poprzedzać
wystąpienie objawów ruchowych lub dołączać się do nich na późniejszym etapie. Część chorych ma objawy typowe dla otępienia czołowo-skroniowego (FTD – frontotemporal dementia), niewielka zaś grupa cechy otępienia o innym profilu (SLA-D – SLA-dementia).
Obraz kliniczny i profile zaburzeń ruchowych w chorobie neuronu ruchowego
Różnorodność fenotypowa choroby neuronu ruchowego (MND – motor neuron disease) jest istotna i koreluje z ciężkością przebiegu choroby, tempem narastania objawów, a także wpływa na sprawność chorego i stopień jego niezależności. Zmienność fenotypu MND wyraża się wiekiem wystąpienia pierwszych objawów, stroną objawów początkowych (przy najczęstszym asymetrycznym początku), stopniem zajęcia górnego bądź dolnego motoneuronu, stopniem progresji objawów i przeżywalnością chorych 1 . Z uwagi na przewagę zajęcia górnego bądź dolnego motoneuronu wyróżniamy postaci: pierwotne stwardnienie boczne (PLS – primary lateral sclerosis), w którym dominuje uszkodzenie (bądź izolowane zajęcie) górnego neuronu ruchowego; postępujący zanik mięśni (PMA – progressive muscular atrophy) z dominującym uszkodzeniem dolnego neuronu ruchowego oraz zespół, w którym współwystępują (w różnym stopniu) cechy uszkodzenia obu grup motoneuronów, określany zbiorczo jako stwardnienie zanikowe boczne. Znaczenie mają również lokalizacja anatomiczna pierwszych i dominujących objawów, jak w postępującym zespole opuszkowym (PBP – progressive bulbar palsy), asymetria objawów, jak w amiotrofii monomelicznej (MMA – monomelic amyotrophy), lub ich symetryczny rozkład, jak w zespole rąk cepowatych 1 .
Diagnostyka neurologiczna i rozpoznanie MND (możliwe, prawdopodobne i pewne) następują na podstawie kryteriów El Escorial, które bazują na obrazie klinicznym i wskaźnikach neurofizjologicznych. Próbą rewizji kryteriów El Escorial są kryteria Awaji, oparte na konsensusie z 2006 r., które proponują dwie fundamentalne zmiany: zastosowanie wyniku badania elektromiograficznego (EMG) i objawów klinicznych do wykazania cech uszkodzenia dolnego motoneuronu (LMN – lower motor neuron) oraz uwzględnienie fascykulacji na równi z fibrylacjami jako markerów aktywnego odnerwienia 1 . Diagnostyka zaburzeń poznawczych i otępienia w przebiegu SLA nie znajduje odzwierciedlenia w kryteriach El Escorial ani Awaji, choć te drugie wskazują na możliwość występowania zaburzeń poznawczych, bez konieczności ich obecności jako kryterium diagnostycznego 2 . Ponadto nie ma jednoznacznych markerów biologicznych otępienia czołowo-skroniowego w przebiegu SLA mimo wielu badań i ich coraz bardziej obiecujących rezultatów. W badaniach uwzględnia się markery z krwi i płynu mózgowo-rdzeniowego, w tym neurofilamenty Nf-H, białko tau i fosforylowane białko tau, TDP-43, ApoE e2 i β-amyloid 1 .
Zaburzenia poznawcze i językowe w SLA
Stwardnienie zanikowe boczne jest postępującą chorobą neurozwyrodnieniową charakteryzującą się zajęciem górnego i dolnego neuronu ruchowego. Częstość występowania zaburzeń poznawczych w SLA szacuje się na 30-50%, choć w niektórych badaniach wskazuje się nawet na ponad 80% 3 . U pacjentów z SLA częste są deficyty wykonawcze i językowe, natomiast funkcje wzrokowo-przestrzenne i pamięć epizodyczna są dobrze zachowane przez długi czas. Wśród zaburzeń funkcji wykonawczych obserwowano różnorodne objawy, takie jak trudności z inicjowaniem czynności, z planowaniem działań wieloetapowych, elastycznością myślenia i kontrolą poznawczą. Ponadto u tych chorych często obserwuje się zaburzenia pamięci operacyjnej 4 .
Obraz kliniczny zaburzeń poznawczych różni się w zależności od postaci SLA. W PLS zaburzenia o profilu otępienia czołowo-skroniowego są częstsze, co może wiązać się z przewagą korowego patomechanizmu tego wariantu MND. W przypadku izolowanego zajęcia wyłącznie dolnego motoneuronu, jak w PMA, znacznie rzadszej postaci MND, częstość występowania zaburzeń poznawczych jest zdecydowanie mniejsza – w jednym z badań z udziałem 12 pacjentów w ogóle ich nie opisano, jednak w późniejszych badaniach wykazano odchylenia w testach badających uwagę i pamięć operacyjną oraz fluencję słowną, szacując ją (mimo niewielkiej grupy badanych) na ok. 17%. W rzadko opisywanym zespole Milla, który jest połowiczą wstępującą bądź zstępującą hemiplegią i może mieć podłoże o charakterze MND, opisywano zaburzenia poznawcze powiązane lokalizacyjnie z uszkodzoną półkulą oraz otępienie.
Komunikacja językowa w SLA ulega zaburzeniom w wyniku działania wielu czynników. Poza dyzartrią i zaburzeniami oddechowymi u wielu pacjentów obserwuje się deficyty językowe. Nie jest to jedynie pogorszenie wyników prób fluencji słownej, które wiąże się zazwyczaj z deficytem wykonawczym, ale mogą to być również zaburzenia nazywania, trudności z przetwarzaniem zdań (deficyt syntaktyczny) i znaczenia słów (deficyt semantyczny). Niekiedy też komunikacja pisemna z pacjentem staje się niemożliwa z powodu agrafii obejmującej pisanie na klawiaturze 5 . W przypadku postaci SLA z postępującą afazją najczęściej obserwuje się wariant z zaburzoną płynnością mowy, ale możliwe jest również występowanie choroby z wariantem semantycznym 6 .
Zaburzenia zachowania w SLA
Najbardziej typowym dla SLA zaburzeniem zachowania jest apatia. Mimo narastającej niesprawności depresja występuje u chorych znacznie rzadziej. W SLA mogą ujawnić się również inne zaburzenia zachowania z kryteriów diagnostycznych wariantu behawioralnego otępienia czołowo-skroniowego (bvFTD – behavioural variant of frontotemporal dementia) poza apatią. U osób z SLA mogą wystąpić m.in.: impulsywność, rozhamowanie, trudności z planowaniem działania, zaburzenia empatii, a zaburzeniom tym mogą towarzyszyć spadek krytycyzmu i brak świadomości zmiany w zakresie zachowania 7 . Co więcej, w SLA występują też zaburzenia zachowania typowe dla innych chorób neurozwyrodnieniowych, takie jak drażliwość i pobudzenie ruchowe 3 .
Znaczenie oceny funkcji poznawczych w SLA
Poziom i profil funkcjonowania poznawczego pacjenta z SLA może mieć znaczenie prognostyczne, ponieważ u chorych z deficytami wykonawczymi obserwuje się szybszą progresję objawów i krótszy czas przeżycia 3 . Ponadto stan funkcji poznawczych ma znaczenie przy podejmowaniu świadomej decyzji o sztucznej wentylacji.
Kryteria diagnostyczne zespołów klinicznych ze spektrum SLA i otępienia czołowo-skroniowego
Rozpoznanie kliniczne SLA ustala się na podstawie wyników badania neurologicznego i oceny neurofizjologicznej. Aktualne kryteria El Escorial i Awaji, pomijające objawy pozaruchowe, spotykają się niekiedy z krytyką. Niektórzy autorzy sugerują włączenie objawów neuropsychologicznych i neuropsychiatrycznych do podstawowych kryteriów diagnostycznych SLA 8 . Z drugiej strony część autorów podkreśla, że dynamika zaburzeń poznawczych w SLA jest bardzo zróżnicowana i ich progresja u pacjentów z dominującymi zaburzeniami ruchowymi jest znacznie wolniejsza niż u osób, u których nakładają się objawy SLA i FTD 9 . Najnowsza propozycja rewizji kryteriów SLA zawiera odniesienie do funkcji poznawczych wskazujące, że zaburzenia poznawcze i zaburzenia zachowania mogą występować w SLA, ale nie są niezbędne do rozpoznania choroby 2 .
Ponieważ u pacjentów z SLA mogą wystąpić objawy typowe dla bvFTD czy afazji pierwotnej postępującej (PPA – primary progressive aphasia), a z kolei u chorych z FTD obserwuje się niekiedy objawy choroby motoneuronu, podjęto kilka prób systematyzacji objawów zespołu, który w starszym piśmiennictwie nazywano FTD z chorobą neuronu ruchowego (FTD-MND – FTD with motor neuron disease) lub otępieniem w SLA (SLA-D). Najnowsze kryteria diagnostyczne systematyzujące spektrum SLA i FTD (SLA-FTSD – frontotemporal spectrum disorder of SLA) opracowane w 2017 r. przez Stronga i wsp. 1 zostały przedstawione całościowo w tabeli 1. Nowe kryteria charakteryzują się wyższą czułością niż poprzednie, opracowane 2009 r. 10 W nowych kryteriach diagnostycznych zoperacjonalizowano kryteria rozpoznawania zaburzeń poznawczych i zaburzeń zachowania typowych dla SLA oraz kryteria rozpoznawania otępienia (SLA-FTD). Wprowadzono też nową kategorię diagnostyczną, obejmującą zarówno zaburzenia poznawcze, jak i zaburzenia zachowania, ale bez otępienia (tab. 1).
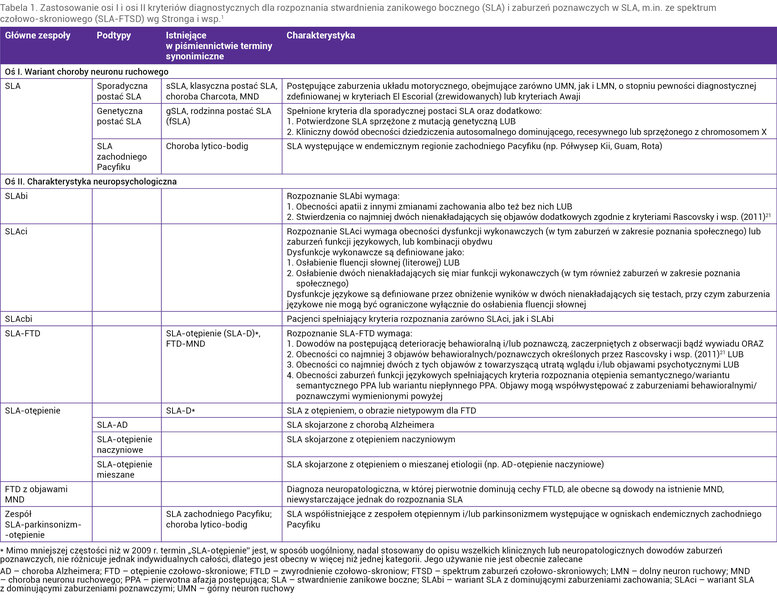
Tabela 1. Zastosowanie osi I i osi II kryteriów diagnostycznych dla rozpoznania stwardnienia zanikowego bocznego (SLA) i zaburzeń poznawczych w SLA, m.in. ze spektrum czołowo-skroniowego (SLA-FTSD) wg Stronga i wsp.1
Na podstawie włoskiej analizy danych klinicznych 797 pacjentów wykazano, że u 48,2% chorych nie stwierdzono zaburzeń poznawczych, 20,5% spełniało kryteria SLA-FTD, 4,8% miało zarówno zaburzenia poznawcze, jak i zaburzenia zachowania (SLAcbi – SLA with cognitive and behavioural impairment), 16,6% ujawniało izolowane zaburzenia poznawcze (SLAci – SLA with cognitive impairment), zaś 7,9% – izolowane zaburzenia zachowania (SLAbi – SLA with behavioral impairment). U 2% osób wykazano obecność zaburzeń poznawczych innych niż dysfunkcje wykonawcze 11 .
Badanie neuropsychologiczne pacjenta z SLA
W ocenie funkcji poznawczych chorego z SLA niezbędne jest uwzględnienie tych obszarów funkcjonowania poznawczego, w których spodziewamy się deficytów (ocena funkcji wykonawczych i poznania społecznego, pamięci operacyjnej i kompetencji językowej), oraz tych, w których zaburzenia obserwuje się w SLA rzadko (ocena pamięci epizodycznej, ocena funkcji wzrokowo-przestrzennych, praksji, jeśli sprawność motoryczna na to pozwala).
Dobór metod diagnostycznych wymaga uwzględnienia profilu objawów ruchowych i aktualnego stanu klinicznego pacjenta 3 . W przypadku niedowładu kończyn górnych zazwyczaj niemożliwe jest wykonanie prób wymagających reakcji motorycznej, co ogranicza zarówno metodologię oceny funkcji wzrokowo-przestrzennych (rezygnacja z oceny zdolności wzrokowo-konstrukcyjnych), jak i pamięci operacyjnej (odstąpienie od prób z ograniczeniem czasowym, wymagających reakcji motorycznej, takich jak Test Łączenia Punktów) oraz pamięci wzrokowej (rezygnacja z prób rysowania czy też układania wzorów). W przypadku występowania anartrii, ale bez znaczącego niedowładu, niekiedy pacjent udziela w badaniu odpowiedzi w formie pisemnej, wykorzystując pismo odręczne, pisanie na klawiaturze tradycyjnej lub z użyciem programu komputerowego, który śledzi ruchy gałek ocznych.
Najłatwiejszym sposobem udzielania odpowiedzi dla pacjenta z głębokimi zaburzeniami ruchowymi są testy wielokrotnego wyboru. Niewiele testów neuropsychologicznych jednak w formie oryginalnej zostało skonstruowanych w tym formacie. Przykładami takich przyjaznych dla pacjenta z SLA zadań są np. podtest Skojarzenia Semantyczne w Sydney Language Battery, oceniający pamięć semantyczną, Test Pamięci Krótkotrwałej Wzrokowej Bentona (BVRT) w wersjach F oraz G czy też Test Matryc Ravena, oceniający zdolność wnioskowania poprzez analogię na materiale wzrokowym.
W przypadku niektórych metod diagnostycznych możliwa jest ich adaptacja umożliwiająca pacjentowi udzielanie odpowiedzi w sposób, który będzie dla niego dostępny. Na przykład próba nazywania konfrontacyjnego może być przeprowadzona zarówno w formie ustnej, jak i pisemnej. Przykładem bardzo udanej adaptacji zadania dla pacjenta z SLA, co więcej, umożliwiającej zróżnicowanie wpływu czynników poznawczych i motorycznych na wynik, jest metodologia oceny fluencji słownej zaproponowana przez Sharon Abrahams i włączona do Edinburgh Cognitive and Behavioural ALS Screen (ECAS; https://ecas.psy.ed.ac.uk). Abrahams zaproponowała możliwość wykonania próby ustnie lub pisemnie, a następnie skorygowanie wyniku względem czasu wykonania próby odczytywania słów (wariant ustny) lub pisania pod dyktando (wariant pisemny). Pozwala to na uniezależnienie wyniku od tempa artykulacji lub pisania 12 .
Niektóre metody badania, na przykład próby uczenia się materiału słownego, są trudne do zaadaptowania dla osoby, która miałaby udzielać odpowiedzi pisemnej, gdyż w takiej formie wymieniany przez pacjenta pisemnie materiał pozostawałby w polu jego widzenia w toku odtwarzania, co mogłoby wpłynąć na wyniki. W przypadku krótkich prób uczenia (np. zapamiętywanie adresu z Addenbrooke’s Cognitive Examination III) jedna z autorek pracy wykorzystywała na przykład pisanie na klawiaturze odtwarzanych słów, ale z wykorzystaniem białej czcionki na białym tle. Taka procedura ma oczywiście charakter eksperymentu i nie wydaje się, aby mogła zastąpić tradycyjne sposoby odtwarzania w przypadku standardowych prób uczenia się materiału słownego.
Aby rozpoznać SLAci u pacjenta z diagnozą SLA, niezbędne jest wykazanie deficytów wykonawczych lub językowych. W przypadku dysfunkcji wykonawczych można je udokumentować na podstawie słabego wyniku próby fluencji literowej lub też słabych wyników dwóch niezależnych prób oceniających funkcje wykonawcze. Analogicznie, w przypadku istotnych zaburzeń językowych ich potwierdzeniem są wyniki dwóch niezależnych testów oceniających te funkcje. Wybiórcze obniżenie fluencji słownej nie stanowi podstawy do rozpoznania zaburzeń językowych w SLA, gdyż może wynikać z deficytów wykonawczych. Na przykład aby wykazać zaburzenia językowe w SLA, zgodnie z kryteriami diagnostycznymi, można by zastosować test nazywania i skojarzeń semantycznych (taki jak z Sydney Language Battery).
W przyszłości być może szersze wykorzystanie technologii śledzenia ruchu gałek ocznych umożliwi wprowadzenie do powszechnej praktyki klinicznej metod oceny, które nie będą wymagały od pacjenta mówienia ani pisania z zaangażowaniem motoryki 3, 13 .
Najczęściej zalecanym narzędziem przesiewowej oceny funkcji poznawczych w SLA jest wspomniana już powyżej skala ECAS. Zasadniczo takie narzędzie nie powinno zawierać prób wymagających reakcji motorycznej 4 . W przypadku diagnozy pacjenta z mniej nasilonymi objawami zajęcia dolnego neuronu ruchowego i braku dostępu do neuropsychologa niekiedy rekomendowane jest wykorzystanie skali Frontal Assessment Battery (FAB), która pozwala na przesiewową ocenę w kierunku deficytów wykonawczych 3 .
Ocena zaburzeń zachowania w SLA
Istnieje wiele skal opracowanych pod kątem wykrywania zaburzeń zachowania w SLA, ale żadna z nich nie jest powszechnie wykorzystywana w praktyce klinicznej. Przeprowadzając wywiad w kierunku zaburzeń zachowania z pacjentem i wiarygodnym informatorem, należy zwrócić szczególną uwagę na objawy bvFTD z kryteriów diagnostycznych oraz na symptomy psychotyczne. Dodatkowo w wywiadzie warto uwzględnić zmianę wrażliwości na bodźce bólowe i nadwrażliwość sensoryczną, spadek krytycyzmu, drażliwość, bladość afektywną, zachowania perseweracyjne (zarówno proste, jak i złożone), zbieractwo, echolalię i zachowanie użytkownika (utilization behaviour; zachowanie wskazujące na zależność od otoczenia, które nie jest inicjowane w sposób wolicjonalny, ale wywołane bodźcem pojawiającym się w zasięgu wzroku/ręki osoby chorej, np. odczytuje ona treść plakietki lekarza czy napis na ścianie, bierze do ręki długopis i zaczyna pisać, sięga po filiżankę i zaczyna pić, nawet jeśli dopiero się napiła i nie jest to jej filiżanka; objaw ten wskazuje na znaczący deficyt hamowania). Okresem porównawczym dla zmiany zachowania może być 10 lat przed pojawieniem się objawów ruchowych, tak jak w Beaumont Behavioural Questionnaire, który jest narzędziem kompleksowo oceniającym potencjalne zmiany zachowania w SLA 14 .
Znaczenie badania genetycznego w diagnostyce różnicowej SLA-FTSD
W ostatnich latach dokonał się znaczny postęp w zrozumieniu podłoża genetycznego SLA i FTD. Nakładające się fenotypy obu chorób związane są szczególnie z mutacjami w genach C9orf72, SQSTM1, VCP, FUS, TBK1. W Europie najczęstszą przyczyną rodzinnej postaci SLA (fSLA – familial SLA) i rodzinnej FTD (fFTD – familial FTD) jest zwielokrotnienie powtórzeń w genie C9orf72 odpowiadające za ok. 40% fSLA i 25% fFTD 15 . Związane z tą mutacją SLA-FTD dziedziczy się autosomalnie dominująco, jednak ma ona niepełną penetrację. Niektóre mutacje związane są przede wszystkim z SLA i rzadko występują w FTD. Są to SOD1, FUS i warianty TARDBP. Podobnie mutacje w genie dla progranuliny częste w FTD nie występują w SLA 16 .
Wiedza na temat podłoża genetycznego obu chorób może być pomocna przy próbie odpowiedzi na tak często zadawane przez pacjentów pytania: dlaczego ja, czy moje dzieci są także zagrożone chorobą, jak szybki będzie postęp choroby? Diagnostyka genetyczna jest jednak trudno dostępna mimo zapotrzebowania ze strony pacjentów i lekarzy. Analiza zachorowań na SLA, FTD, inne otępienia, choroby psychiatryczne, parkinsonizm czy samobójstw w rodzinie (co najmniej trzech pokoleń) może sugerować sposób dziedziczenia choroby, na przykład transmisja autosomalna dominująca jest stosunkowo częsta w SLA. Niestety ograniczona penetracja genu, brak informacji o rodzinie, błędne rozpoznania, wczesny zgon, nieujawniona adopcja i inne okoliczności mogą doprowadzić do błędnych wniosków. Prawdopodobieństwo nosicielstwa znanej mutacji o wysokiej penetracji w przypadku sporadycznego zachorowania na SLA wynosi 11%. Natomiast przy fenotypie SLA-FTD wynosi już 88% nawet przy ujemnym wywiadzie rodzinnym. W obu przypadkach najczęstszą mutacją jest zwielokrotnienie powtórzeń w genie C9orf72 17 . Wszyscy pacjenci z SLA, którzy mają europejskie korzenie, powinni mieć możliwość przeprowadzenia badania w kierunku mutacji w C9orf72, ponieważ dostępne są już badania kliniczne dla nosicieli tej mutacji. Badania u osób zdrowych powinny być zarezerwowane dla dorosłych krewnych pierwszego stopnia z potwierdzoną mutacją.
Znaczenie badań obrazowych w diagnostyce SLA-FTSD
Badania obrazowe w diagnostyce SLA-FTSD mają charakter pomocniczy i, w przeciwieństwie do bvFTD czy PPA, żaden ze wzorców wyników badań neuroobrazowych nie zwiększa pewności rozpoznania. Badania obejmujące ocenę morfologiczną z zastosowaniem rezonansu magnetycznego: morfometria oparta na wokselach (VBM – voxel-based morphometry) i morfometria oparta na powierzchni (SBM – surface-based morphometry), wykorzystując wysokiej rozdzielczości MR, pozwalają na wykrywanie ogniskowych zmian w obrębie istoty szarej. Technika ta pozwala (poza uwidacznianiem zmian w korze ruchowej, co do których nie ma wątpliwości, biorąc pod uwagę patomechanizm choroby) również na zobrazowanie wieloogniskowych zmian w okolicach czołowej, skroniowej i ciemieniowej 18 . Zmiany ogniskowe opisywane były też w strukturach podkorowych, w tym w hipokampie, ciele migdałowatym, wzgórzu i wyspie 19 . Rozmieszczenie zmian korowych w strukturach pozaruchowych potwierdza kontinuum SLA-FTSD.
Ocena istoty białej w MR oparta jest przede wszystkim na technice obrazowania tensora dyfuzji (DTI – diffusion tensor imaging), w której zredukowane parametry anizotropii oraz zwiększona dyfuzja w drogach korowo-rdzeniowych i w obrębie ciała modzelowatego są charakterystycznymi wskaźnikami w badaniu chorych z SLA. Zmiany w istocie białej związane z objawami pozaruchowymi opisywane są natomiast w okolicach czołowych, skroniowych, w korze obręczy, wyspy, wzgórza i w móżdżku 19 .
Kolejna grupa badań diagnostycznych obejmuje techniki obrazowania metabolizmu – spektroskopię MR (w której charakterystyczne są obniżone wskaźniki NAA/cholina i NAA/kreatyna w okolicach odpowiedzialnych za ruch, ale również w płatach czołowych, ciemieniowych, we wzgórzu i potylicy); pozytonową tomografię emisyjną ze znacznikiem 18 F-fludeoksyglukozą ( 18 F-FDG PET – 18-F-FDG positron emission tomography), która wykazuje hipometabolizm, zwłaszcza w okolicach grzbietowo-bocznych kory przedczołowej, zakrętach oczodołowych, przedniej okolicy czołowej i skroniowej, zakręcie wrzecionowatym, a także w korze potylicznej 20 . Warto zauważyć, że wykazany hipometabolizm, korelujący z zaburzeniami poznawczymi, wyprzedza zwykle zanik tkanki mózgowej możliwy do zaobserwowania w badaniach morfologicznych 19 . PET pozwala również na zobrazowanie aktywacji mikrogleju w okolicach czołowo-skroniowych, wzgórzu, śródmózgowiu i moście, co wskazuje na proces zapalny 19 .
Znaczenie kliniczne zaburzeń poznawczych w SLA
Wielu neurologów, psychologów, fizjoterapeutów, neurologopedów, lekarzy rodzinnych oraz innych specjalistów zajmujących się opieką nad pacjentami z SLA nie jest świadomych współistnienia zaburzeń poznawczych i zaburzeń zachowania z objawami ruchowymi. Wpływa to na brak zrozumienia zgłaszanych przez chorych i/lub opiekunów dolegliwości. Brak uwzględnienia w pracy z pacjentami z SLA dysfunkcji poznawczych i zaburzeń zachowania zmniejsza skuteczność ich rehabilitacji ruchowej i neurologopedycznej. Współwystępowanie zaburzeń funkcji poznawczych i otępienia silnie koreluje z gorszym rokowaniem, wyższą śmiertelnością, szybszą progresją objawów ruchowych (w tym zwłaszcza opuszkowych) i gorszą postacią kliniczną choroby. Stwierdzenie u chorego otępienia wyklucza jego możliwości świadomego decydowania o swoim losie, w tym o ewentualnej wentylacji mechanicznej. Zaburzenia językowe mogą powodować trudności w rozumieniu złożonych kwestii, formularzy zgody, umów itp. Badania nad zaburzeniami funkcji poznawczych i zachowania, ich profilem oraz częstością występowania otępienia, w tym zwłaszcza otępienia czołowo-skroniowego w SLA, mają ogromne znaczenie w zrozumieniu patomechanizmu tych schorzeń i przybliżają nas do znalezienia skutecznych metod ich leczenia.
Podsumowanie
Pacjenci z SLA wymagają monitorowania stanu poznawczego i emocjonalnego. Dysfunkcje poznawcze i zaburzenia zachowania w istotnym stopniu przyczyniają się do niesprawności wielu chorych z SLA. Rozpoznanie SLA-FTSD nie tylko ma znaczenie z punktu widzenia oddziaływań psychoedukacyjnych kierowanych do rodziny pacjenta, ale też powinno przekładać się na odpowiednie dostosowanie oddziaływań fizjoterapeutycznych i neurologopedycznych. Kluczowe znaczenie dla dowolnego terapeuty pracującego z pacjentem ma zrozumienie faktu, że apatia, która może objawiać się obniżoną motywacją do ćwiczeń, jest objawem dysfunkcji mózgowej. W przypadku pacjenta z apatią gotowość do samodzielnego powtarzania zaleconych ćwiczeń może być znacząco ograniczona.
Abstract
Disorders due to amyotrophic lateral sclerosis and frontotemporal dementia: diagnostic criteria and differential diagnosis
About 70% of amyotrophic lateral sclerosis (ALS) patients suffer cognitive impairments. It should be noted that 30-50% of people with ALS and other forms of motor neuron disease are diagnosed with cognitive, language and behavioral disturbances that correspond to frontotemporal dementia (FTD) with predominant behavioral, executive or language impairment. Visuospatial functions and episodic memory are well preserved. Behavioral manifestations are dominated by apathy as well as other changes associated with the spectrum of behavioral variant FTD, including impulsiveness, disinhibition, empathy disorders, decreased criticism, irritability and agitation. Communication disorders associated with ALS are attributable to a number of factors: dysarthria and respiratory dysfunction as well as language deficits. Non-fluent aphasia and/or agraphia may also be present. This paper presents diagnostic criteria for amyotrophic lateral sclerosis – frontotemporal spectrum disorder (ALS-FTSD) as developed in 2017. A neuropsychological assessment should include methods focused on the expected profile of cognitive and behavioral deficits while taking into account the patient’s motor problems. There is a growing understanding of the genetic causes of ALS and ALS-FTSD, and increasingly more genetic tests are available, among which testing for multiplication of repeats in the C9orf72 gene is the most clinically useful in Europe. The identification and understanding of cognitive impairments in ALS patients come with a significant prognostic value as well as facilitate physiotherapy and speech rehabilitation. This is also important when the patient makes important life choices, e.g. decides whether to give consent to mechanical ventilation.
- 1. Strong MJ, Abrahams S, Goldstein LH, et al. Amyotrophic lateral sclerosis – frontotemporal spectrum disorder (ALS-FTSD): Revised diagnostic criteria. Amyotroph Lateral Scler Frontotemporal Degener 2017;18(3-4):153-74
- 2. Shefner JM, Al-Chalabi A, Baker MR, et al. A proposal for new diagnostic criteria for ALS. Clin Neurophysiol 2020;131(8):1975-8
- 3. Huynh W, Ahmed R, Mahoney CJ, et al. The impact of cognitive and behavioral impairment in amyotrophic lateral sclerosis. Expert Rev Neurother 2020;20(3):281-93
- 4. Goldstein LH, Abrahams S. Changes in cognition and behaviour in amyotrophic lateral sclerosis: nature of impairment and implications for assessment. Lancet Neurol 2013;12(4):368-80
- 5. Sitek E, Kluj-Kozłowska K, Narożańska E. Nietypowe zaburzenia mowy i pisma jako wczesne objawy choroby neurozwyrodnieniowej. Neurol Dypl 2021;16(2):42-50
- 6. Tan RH, Guennewig B, Dobson-Stone C, et al. The underacknowledged PPA-ALS: A unique clinicopathologic subtype with strong heritability. Neurology 2019;92(12):e1354-e1366
- 7. Benbrika S, Desgranges B, Eustache F, Viader F. Cognitive, Emotional and Psychological Manifestations in Amyotrophic Lateral Sclerosis at Baseline and Overtime: A Review. Front Neurosci 2019;13:951
- 8. Al-Chalabi A, Hardiman O, Kiernan MC, Chiò A, Rix-Brooks B, van den Berg LH. Amyotrophic lateral sclerosis: moving towards a new classification system. Lancet Neurol 2016;15(11):1182-94
- 9. Lulé DE, Aho-Özhan HEA, Vázquez C, et al. Story of the ALS-FTD continuum retold: rather two distinct entities. J Neurol Neurosurg Psychiatry 2019;90(5):586-9
- 10. Iazzolino B, Pain D, Peotta L, et al. Validation of the revised classification of cognitive and behavioural impairment in ALS. J Neurol Neurosurg Psychiatry 2019;90(7):734-9
- 11. Chiò A, Moglia C, Canosa A, et al. Cognitive impairment across ALS clinical stages in a population-based cohort. Neurology 2019;93(10):e984-e994
- 12. Abrahams S, Newton J, Niven E, Foley J, Bak TH. Screening for cognition and behaviour changes in ALS. Amyotroph Lateral Scler Frontotemporal Degener 2014;15(1-2):9-14
- 13. Bueno APA, Sato JR, Hornberger M. Eye tracking – The overlooked method to measure cognition in neurodegeneration? Neuropsychologia 2019;133:107191
- 14. Gosselt IK, Nijboer TCW, van Es MA. An overview of screening instruments for cognition and behavior in patients with ALS: selecting the appropriate tool for clinical practice. Amyotroph Lateral Scler Frontotemporal Degener. 2020;1-13. Published online ahead of print 2020 Mar 11
- 15. Ranganathan R, Haque S, Coley K, et al. Multifaceted genes in amyotrophic lateral sclerosis-frontotemporal dementia. Front Neurosci 2020,14:684
- 16. Abramzon YA, Fratta P, Traynor BJ, et al. The overlapping genetics of Amyotrophic lateral sclerosis and frontotemporal dementia. Front Neurosci 2020;14:42
- 17. Roggenbuck J, Quick A, Kolb SJ. Genetic testing and genetic counseling for amyotrophic lateral sclerosis: an update for clinicians. Genet Med 2017;19(3):267-74
- 18. Crespi C, Dodich A, Cappa SF, et al. Multimodal MRI quantification of the common neurostructural bases within the FTD-ALS continuum, Neurobiol Aging 2018;62:95-104
- 19. Christidi F, Karavasilis E, Rentzos M, et al. Clinical and radiological markers of extra-motor deficits in Amyotrophic lateral sclerosis. Front Neurol 2018;9:1005
- 20. Rajagoplan V, Pioro E. 2‐Deoxy‐2‐[18F]fluoro‐d‐glucose Positron Emission Tomography, cortical thickness and white matter graph network abnormalities in brains of patients with Amyotrophic lateral sclerosis and frontotemporal dementia suggest early neuronopathy rather than axonopathy. Eur J Neurol 2020;10.1111/ene.14332. doi: 10.1111/ene.14332. Published online ahead of print 2020 May 20
- 21. Kuklińska M, Sitek EJ, Brockhuis i wsp. Wariant behawioralny otępienia czołowo-skroniowego – wybrane problemy diagnostyczne w neuropsychiatrii. Aktualn Neurol 2020;20(2): 71-81




 Dodaj do ulubionych
Dodaj do ulubionych
