


Co znajdziesz w artykule?
- W diagnostyce różnicowej choroby neuronu ruchowego i chorób imitujących można zastosować podział uwzględniający obecność objawów towarzyszących, takich jak zaburzenia czucia czy drżenie kończyn, a także dystrybucję niedowładu
- W artykule opisano wybrane jednostki chorobowe, które wymagają różnicowania ze stwardnieniem zanikowym bocznym, niezależnie od etiologii
Spis treści
Choroby neuronu ruchowego (MND – motor neurone disease) to grupa schorzeń o szerokim zakresie objawów klinicznych i zróżnicowanej etiologii. Ich wspólną cechą jest niedowład, który wynika z uszkodzenia drogi ruchowej. Stwardnienie boczne zanikowe (ALS – amyotrophic lateral sclerosis) jest najczęstszą chorobą neuronu ruchowego. Pozostałe choroby motoneuronu odpowiadają za około 15% rozpoznań. W klasycznej postaci ALS występuje jednoczesne uszkodzenie górnego i dolnego neuronu ruchowego. W
większości pozostałych chorób motoneuronu dominuje uszkodzenie dolnego neuronu ruchowego na poziomie jąder ruchowych nerwów czaszkowych opuszki, komórek ruchowych rogów przednich rdzenia kręgowego lub włókien ruchowych nerwów obwodowych. Objawy chorób górnego neuronu ruchowego wynikają z uszkodzenia neuronów kory ruchowej oraz dróg korowo-rdzeniowych i/lub korowo-opuszkowych (tab. 1).

Tabela 1. Podział chorób neuronu ruchowego (MND) w zależności od miejsca uszkodzenia
Definicja MND nie jest sprecyzowana, a dane zawarte w piśmiennictwie są sprzeczne. Niektórzy autorzy zaliczają do MND jedynie choroby o etiologii neurodegeneracyjnej, a wszystkie inne schorzenia z podobnymi objawami klinicznymi określają jako choroby imitujące MND. Inni z kolei w klasyfikacji MND uwzględniają także choroby uwarunkowane genetycznie oraz nabyte, w tym o podłożu autoimmunologicznym. W artykule opisano wybrane jednostki chorobowe, które wymagają różnicowania z ALS, niezależnie od etiologii.
W diagnostyce różnicowej MND i chorób imitujących przydatny jest podział chorób uwzględniający obecność objawów towarzyszących, takich jak zaburzenia czucia czy drżenie kończyn, oraz dystrybucję niedowładu. Niedowład w chorobach z kręgu MND może mieć rozkład symetryczny bądź asymetryczny, być zlokalizowany proksymalnie lub dystalnie (tab. 2).

Tabela 2. Podział MND w zależności od lokalizacji niedowładu
Wstępne rozpoznanie MND ustala się na podstawie objawów klinicznych. W dalszej diagnostyce większości schorzeń niezbędne są badanie przewodnictwa nerwowego i elektromiograficzne oraz obrazowanie, które pozwalają określić charakter i poziom uszkodzenia neuronu ruchowego.
Nabyte choroby neuronu ruchowego
Wieloogniskowa neuropatia ruchowa z blokiem przewodzenia
Wieloogniskowa neuropatia ruchowa z blokiem przewodzenia (MMN – multifocal motor neuropathy) jest neuropatią czysto ruchową, która występuje około 10 razy rzadziej niż ALS. Do wystąpienia choroby dochodzi z reguły przed 50 rokiem życia (średni wiek zachorowania to 40 lat). Mężczyźni chorują około 3 razy częściej niż kobiety.
Rozwój choroby ma związek z obecnością przeciwciał IgM skierowanych przeciwko gangliozydom GM1, które są zlokalizowane w obrębie włókien ruchowych w okolicy przewężeń Ranviera oraz paranodalnie. Reakcja autoimmunologiczna powoduje demielinizacyjne uszkodzenie nerwów obwodowych. Obecność przeciwciał w wysokim mianie stwierdza się u około 50-60% pacjentów z MMN. Przeciwciała te nie są specyficzne dla tej neuropatii, można je również wykryć u 5-10% chorych na ALS, zazwyczaj jednak w niskim mianie.
Choroba postępuje powoli lub skokowo. W MMN dochodzi do asymetrycznego i wieloogniskowego uszkodzenia nerwów obwodowych. W 2/3 przypadków objawy rozwijają się w obrębie kończyn górnych, głównie dystalnie. W 1/3 przypadków choroba zaczyna się od opadania stopy. Tylko u 5% pacjentów pierwszym objawem choroby jest proksymalny niedowład kończyny górnej.
W odróżnieniu od ALS, w którym dochodzi do uszkodzenia całego segmentu rdzenia kręgowego (niedowład mięśni wynika z uszkodzenia miotomu), MMN charakteryzuje się uszkodzeniem pojedynczych nerwów obwodowych. Innymi cechami odróżniającymi MMN od ALS są rozkład niedowładu w kończynach górnych i zanik mięśni. Typowym objawem w MMN jest opadanie palców i/lub ręki wynikające z dysfunkcji prostowników przedramienia. W ALS natomiast pierwszymi zajętymi mięśniami są najczęściej mięśnie kłębu i międzykostne. W MMN zwraca uwagę stosunkowo nieduży zanik mięśni dotkniętych niedowładem, z kolei w ALS od początku choroby obserwuje się wyraźny zanik mięśni.
U 50% chorych na MMN występują nasilone kurcze mięśni oraz fascykulacje. Odruchy głębokie są z reguły osłabione. Jedynie w 20% przypadków mogą być żywe, a u 8% chorych nawet wygórowane. W przebiegu choroby nie dochodzi do zajęcia włókien czuciowych, ale pacjenci mogą zgłaszać niewielkie zaburzenia czucia wibracji w obrębie kończyn dolnych. W MMN nie występują objawy opuszkowe.
Rozpoznanie choroby ustala się na podstawie objawów klinicznych, wyniku badania elektrofizjologicznego oraz wyników badań dodatkowych, takich jak rezonans magnetyczny splotów ramiennych i badanie ogólne płynu mózgowo-rdzeniowego. Kryteria kliniczne dla MMN przedstawiono w tabeli 3.

Tabela 3. Kryteria kliniczne rozpoznania wieloogniskowej neuropatii ruchowej z blokiem przewodzenia (MMN) wg European Federation of Neurological Societies/Peripheral Nerve Society Guideline on Management of Multifocal Motor Neuropathy 2010
Kluczową rolę w diagnostyce MMN odgrywa badanie elektrofizjologiczne. Jego celem jest wykazanie bloku przewodzenia we włóknach ruchowych nerwów obwodowych w miejscach nienarażonych na ucisk. Występowanie bloku przewodzenia wynika z odcinkowego uszkodzenia mieliny we włóknach ruchowych i braku możliwości propagacji potencjału wzdłuż nieuszkodzonego aksonu. Przewodnictwo czuciowe w MMN jest prawidłowe.
W diagnostyce choroby przydatny jest rezonans magnetyczny splotów ramiennych. W 40-50% przypadków stwierdza się hiperintensywność splotu w sekwencji T2, pogrubienie pęczków oraz wzmocnienie pokontrastowe splotu. Opisywane zmiany są asymetryczne.
W badaniu ogólnym płynu mózgowo-rdzeniowego u 30% chorych na MMN stężenie białka jest umiarkowanie zwiększone (<1 g/l).
MMN jest neuropatią poddającą się leczeniu, dlatego tak ważne jest wczesne rozpoznanie choroby. Niestety średni czas od wystąpienia pierwszych objawów do ustalenia rozpoznania wynosi 4 lata. Lekiem pierwszego rzutu są immunoglobuliny podawane w dawce 2 g/kg m.c. przez 5 dni co 2-6 tygodni. W miarę postępu choroby zmniejsza się odpowiedź na immunoglobuliny, co wynika z wtórnego uszkodzenia aksonów nerwów obwodowych.
Amiotrofia monomeliczna
Amiotrofia monomeliczna (MA – monomelic amyotrophy; choroba Hirayamy) jest bardzo rzadką chorobą dolnego neuronu ruchowego. Występuje głównie w Japonii i Indiach. Chorobowość jest nieznana. MA zdecydowanie częściej występuje u mężczyzn (M:K 10:1). Do zachorowania dochodzi około 20 roku życia.
W przebiegu MA następuje ogniskowy zanik i niedowład mięśni jednej kończyny górnej. U części chorych może rozwinąć się mniej nasilony niedowład kończyny przeciwnej. Charakterystyczne jest zajęcie mięśni przedramienia i ręki, które są unerwiane przez korzenie C7-Th1. Typowe jest zaoszczędzenie mięśnia ramienno-promieniowego (korzeń C5-C6). Choroba postępuje powoli, objawy narastają w czasie od roku do 5 lat, a następnie ulegają stabilizacji. Niedowład może być nasilany przez zimno. U części chorych obserwuje się niewielkie drżenie kończyny. W MA nie występują objawy czuciowe, opuszkowe ani piramidowe.
Bardzo rzadko amiotrofia monomeliczna może się rozwinąć w obrębie kończyny dolnej. Niedowład i zanik dotyczą głównie mięśnia brzuchatego łydki i mięśnia płaszczkowatego. W tych przypadkach przebieg choroby jest bardzo łagodny.
Etiologia choroby pozostaje nieznana. Jako przyczynę bierze się pod uwagę naczyniowe uszkodzenie rdzenia kręgowego, wynikające z ucisku naczyń tętniczych przez oponę twardą w trakcie zginania szyi. W badaniu rezonansu magnetycznego kręgosłupa stwierdza się zanik rdzenia kręgowego oraz hiperintensywny sygnał rdzenia na poziomie C5-C7. W badaniach czynnościowych w trakcie zgięcia szyi obserwuje się poszerzenie splotów żylnych opony twardej na grzbietowej powierzchni rdzenia.
W badaniu elektromiograficznym stwierdza się neurogenny zapis z mięśni unerwianych przez korzenie C7-Th1 (lub S1-S2 w przypadku zajęcia kończyn dolnych). Przewodnictwo nerwowe jest prawidłowe.
MA należy różnicować z radikulopatią, mielopatią szyjną oraz z ALS. W przypadku radikulopatii lub mielopatii pomocną rolę w diagnostyce różnicowej odgrywa badanie rezonansu magnetycznego. Z kolei ALS rzadko rozwija się w młodym wieku, jest chorobą postępującą i w jej przebiegu nie dochodzi do wybiórczego niedowładu mięśni.
Ostra ruchowa neuropatia aksonalna
Ostra ruchowa neuropatia aksonalna (AMAN – acute motor axonal neuropathy) jest wariantem zespołu Guillaina-Barrégo (GBS – Guillain-Barré syndrome), który w Europie występuje rzadko, ale jest najczęstszym wariantem choroby występującym w Chinach. Jego cechami charakterystycznymi są: nagły początek, wiotki, wstępujący, symetryczny niedowład kończyn, bardziej nasilony dystalnie, oraz osłabienie lub brak odruchów głębokich.
W 85% przypadków choroba poprzedzona jest infekcją przewodu pokarmowego wywołaną pałeczką Campylobacter jejuni. W mechanizmie mimikry molekularnej między liposacharydami ściany komórkowej bakterii a molekułami znajdującymi się na powierzchni aksonu produkowane są przeciwciała IgG, które są skierowane przeciwko gangliozydom GM1 i Gd1a.
W badaniu przewodnictwa nerwowego stwierdza się cechy uszkodzenia aksonu (obniżone amplitudy odpowiedzi ruchowych). Parametry przewodzenia we włóknach czuciowych są prawidłowe. Zapis elektromiograficzny wskazuje na uszkodzenie neurogenne badanych mięśni z licznymi potencjałami odnerwiennymi.
AMAN lepiej reaguje na leczenie immunoglobulinami niż plazmaferezą.
Czysto ruchowa postać przewlekłej zapalnej polineuropatii demielinizacyjnej
Atypowa, czysto ruchowa postać przewlekłej zapalnej polineuropatii demielinizacyjnej (CIDP – chronic inflammatory demyelinating polyneuropathy) występuje bardzo rzadko i stanowi 7-10% przypadków CIDP. Choroba rozwija się częściej u młodych osób (zazwyczaj przed 20 rokiem życia). Charakteryzuje się symetrycznym, dystalnym i proksymalnym niedowładem kończyn. Objawy czuciowe nie występują lub są bardzo dyskretne.
Przyczyna rozwoju CIDP nie jest dostatecznie poznana. Podejrzewa się udział mechanizmów zarówno komórkowych, jak i humoralnych. W surowicy z reguły nie wykrywa się żadnych przeciwciał. W badaniu przewodnictwa nerwowego stwierdza się uszkodzenie włókien ruchowych nerwów obwodowych o charakterze pierwotnie demielinizacyjnym. Parametry przewodzenia we włóknach czuciowych są prawidłowe.
Lekami pierwszego rzutu w leczeniu CIDP są immunoglobuliny i kortykosteroidy.
Uwarunkowane genetycznie choroby neuronu ruchowego
Rdzeniowy zanik mięśni
Rdzeniowy zanik mięśni (SMA – spinal muscular atrophy) jest najczęstszą genetycznie uwarunkowaną chorobą neuronu ruchowego. Jest schorzeniem dziedziczonym w sposób autosomalnie recesywny. Za rozwój choroby odpowiadają dwa geny: SMN1 i SMN2. Do jej wystąpienia dochodzi w wyniku homozygotycznej delecji (95-98%) lub mutacji punktowej genu SMN1 (gen przeżycia motoneuronu), który jest zlokalizowany na chromosomie 5. Gen ten wykazuje największą ekspresję w neuronach ruchowych. Gen SMN2 odpowiedzialny jest za czas wystąpienia objawów, ich nasilenie i przebieg choroby. Im więcej kopii genu SMN2, tym łagodniejszy jest przebieg choroby.
SMA dzieli się na pięć podtypów wyróżnionych na podstawie wieku wystąpienia objawów choroby i osiągnięcia kamieni milowych rozwoju motorycznego. Początek choroby w wieku dorosłym występuje w typie 4 SMA i u części chorych w typie 3b. Typ 4 stanowi <5% przypadków SMA. Mężczyźni i kobiety chorują z równą częstością. Objawy choroby rozwijają się zazwyczaj po 30 roku życia, a początek ich wystąpienia jest trudny do wychwycenia. Pacjenci zgłaszają pogorszenie sprawności ruchowej i upadki. Mają trudności z wchodzeniem po schodach lub ze wstawaniem z pozycji kucznej. Choroba postępuje powoli. W obrazie klinicznym dominuje proksymalny, symetryczny niedowład, głównie kończyn dolnych, z zanikiem i kurczami mięśni oraz fascykulacjami. Odruchy głębokie są osłabione lub zniesione. Dosyć typowym objawem jest drżenie pozycyjne i kinetyczne kończyn górnych.
U chorych nie występują zaburzenia czucia. Rzadko w przebiegu choroby można zaobserwować przerost mięśni prostowników krótkich palców i mięśni łydek.
W badaniach laboratoryjnych stwierdza się zwiększoną aktywność kinazy kreatynowej. Jest ona podwyższona 2-4 razy powyżej górnej granicy normy, ale z reguły <10.
Z uwagi na zajęcie mięśni obręczy barkowej i biodrowej, przerost mięśni łydek i zwiększoną aktywność kinazy kreatynowej SMA jest często określany jako choroba imitująca miopatię. W takich przypadkach na podstawie badania elektromiograficznego można ją wykluczyć.
W związku z tym, że w SMA zwyrodnieniu ulegają komórki ruchowe rogów przednich rdzenia kręgowego, w zapisie elektromiograficznym stwierdza się uszkodzenie neurogenne badanych mięśni. Zapisy wysiłkowe są zubożone, wysokonapięciowe, nierzadko o amplitudzie wynoszącej >10 mV. Wartości parametrów potencjałów jednostek ruchowych są znacznie zwiększone. W spoczynku w badanych mięśniach rejestruje się fascykulacje i potencjały odnerwienne (fibrylacje i dodatnie fale ostre). Całość badania elektrofizjologicznego wskazuje na toczący się jednocześnie proces de- i reinerwacji. Przewodnictwo ruchowe i czuciowe w SMA są prawidłowe.
Biorąc pod uwagę rozkład niedowładu, w diagnostyce różnicowej SMA należy uwzględnić ALS, głównie pierwotny zanik mięśni. Choroba wymaga również różnicowania z miopatiami, rdzeniowo-opuszkowym zanikiem mięśni (u mężczyzn) i przewlekłą zapalną polineuropatią demielinizacyjną.
Wprowadzenie do leczenia nusinersenu powoduje, że wczesne rozpoznanie choroby daje szanse na uniknięcie niesprawności.
Rdzeniowo-opuszkowy zanik mięśni
Rdzeniowo-opuszkowy zanik mięśni (SBMA – spinal and bulbar muscular atrophy; choroba Kennedy’ego) jest kolejną rzadką chorobą neuronu ruchowego. SBMA jest dziedziczony w sposób recesywny, sprzężony z chromosomem X. U chorych występuje wzrost liczby powtórzeń trinukleotydów CAG w pierwszym eksonie genu dla receptora androgenowego. Im większa jest liczba powtórzeń, tym cięższy przebieg choroby. Dokładny mechanizm prowadzący do rozwoju choroby pozostaje nieznany. W wyniku mutacji dochodzi do agregacji nieprawidłowego białka i tworzenia się wtrętów wewnątrz jądra komórki. Prowadzi to do zaburzeń transkrypcji. Choroba rozwija się tylko u mężczyzn między 30 a 60 rokiem życia. Objawy kliniczne wynikają z uszkodzenia komórek ruchowych rogów przednich rdzenia kręgowego i jąder ruchowych opuszki.

Tabela 4. Objawy sugerujące rdzeniowo-opuszkowy zanik mięśni
Charakterystycznym objawem SBMA jest niedowład proksymalny, początkowo kończyn dolnych, z towarzyszącym bólem i kurczami mięśni. Na początku choroby niedowład może mieć rozkład asymetryczny (około 50% chorych). Choroba rzadko zaczyna się od zajęcia mięśni regionu opuszki. Wraz z postępem SBMA rozwija się dyzartria, dysfagia i dysfonia. Charakterystyczne dla choroby są fascykulacje w okolicy perioralnej i w obrębie języka oraz zanik języka. Do jej objawów należy także drżenie kończyn górnych, które wyprzedza wystąpienie niedowładu kończyn dolnych nawet o dekadę. Połowa chorych ma objawy polineuropatii czuciowej (tab. 4). Kobiety – nosicielki nieprawidłowego genu – mogą się skarżyć na kurcze mięśni.
W SBMA oprócz objawów ruchowych występują także zaburzenia endokrynologiczne. Należą do nich: obniżona płodność, ginekomastia, zaburzenia metabolizmu glukozy lub lipidowe.
Wynik badania elektromiograficznego jest podobny jak w SMA. Ponadto w SBMA stwierdza się cechy uszkodzenia neurogennego z obecnością licznych fascykulacji w mięśniach regionu opuszki. Przewodnictwo czuciowe jest nieprawidłowe. Rejestruje się obniżone amplitudy odpowiedzi czuciowych z nerwów obwodowych.
SBMA wymaga różnicowania przede wszystkim z ALS. W ALS nie stwierdza się drżenia rąk, objawów czuciowych ani zaburzeń endokrynologicznych. Ponadto postęp choroby w ALS jest dużo szybszy. Rozpoznanie SBMA, podobnie jak SMA, ustalane jest na podstawie badania genetycznego.
Dziedziczne neuropatie ruchowe
Dziedziczne neuropatie ruchowe (dHMN – distal hereditary motor neuropathies) to bardzo rzadka i niezwykle heterogenna pod względem genetycznym i klinicznym grupa chorób dolnego neuronu ruchowego, które mogą być dziedziczone w sposób autosomalnie dominujący, autosomalnie recesywny lub sprzężony z chromosomem X. Dotychczas odkryto mutację kilkunastu genów odpowiedzialnych za rozwój choroby. Mutacja jednego genu może powodować różne fenotypy choroby. Szacuje się jednak, że w 80% przypadków dHMN są spowodowane mutacją jeszcze nieodkrytego genu. U większości chorych objawy rozwijają się w ciągu dwóch pierwszych dekad życia (tab. 5).
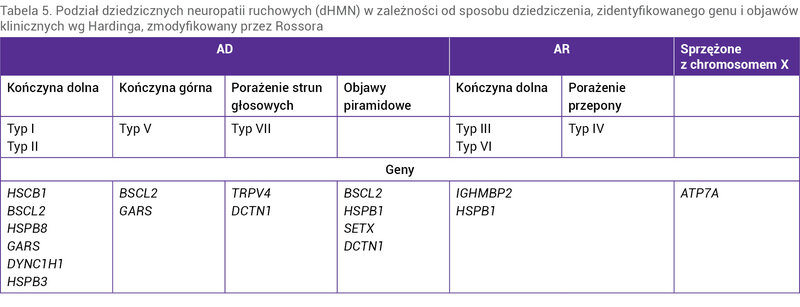
Tabela 5. Podział dziedzicznych neuropatii ruchowych (dHMN) w zależności od sposobu dziedziczenia, zidentyfikowanego genu i objawów klinicznych wg Hardinga, zmodyfikowany przez Rossora
Dziedziczne neuropatie ruchowe charakteryzują się powoli postępującym, symetrycznym i głównie dystalnym niedowładem kończyn dolnych, bez towarzyszących zaburzeń czucia. W dHMN nie występują objawy opuszkowe. Do rozwoju objawów dochodzi w dzieciństwie, a początek ich wystąpienia jest trudny do wychwycenia. Objawy zazwyczaj rozpoczynają się w obrębie stóp (stopa wydrążona, płaska, palce młotkowate), z czasem wstępują. Wyjątkiem jest typ V wg Hardinga, w którym pierwsze objawy rozwijają się w obrębie kończyn górnych.
Oprócz niedowładu kończyn w niektórych typach dHMN występują objawy towarzyszące, np. porażenie przepony lub porażenie strun głosowych (tab. 5).
Istotną rolę w diagnostyce dHMN odgrywa badanie elektrofizjologiczne, które pozwala różnicować tę grupę chorób z polineuropatiami ruchowo-czuciowymi uwarunkowanymi genetycznie (CMT – Charcot-Marie-Tooth disease) oraz z dystalnymi miopatiami. W badaniu przewodnictwa nerwowego stwierdza się obniżone amplitudy odpowiedzi ruchowych; zmiany są zależne od długości włókien. W przeciwieństwie do CMT przewodnictwo czuciowe jest prawidłowe. Badanie elektromiograficzne potwierdza przewlekle neurogenne uszkodzenie mięśni dystalnych.
Rozpoznanie choroby ustala się na podstawie obrazu klinicznego, wyniku badania elektrofizjologicznego i wyniku badania genetycznego.
Dziedziczne paraparezy spastyczne
Dziedziczne paraparezy spastyczne (HSP – hereditary spastic paraplegia) to grupa chorób, w przebiegu których dochodzi do uszkodzenia górnego neuronu ruchowego. Stanowią heterogenną grupę schorzeń, które mogą być dziedziczone w sposób autosomalnie dominujący, autosomalnie recesywny, sprzężony z chromosomem X lub mitochondrialnie. Opisywane są również przypadki sporadyczne. Dotychczas odkryto ponad 70 genów odpowiedzialnych za rozwój choroby (tab. 6).
HSP występują rzadko. Chorobowość wynosi 1,8/100 000. Choroba postępuje powoli. Paraparezy spastyczne dziedziczone w sposób autosomalnie dominujący rozwijają się zazwyczaj w wieku dorosłym (między drugą a trzecią dekadą życia), a dziedziczone w sposób autosomalnie recesywny – w dzieciństwie lub u młodych dorosłych.
Głównym objawem HSP jest spastyczny niedowład kończyn dolnych, który wynika z uszkodzenia dróg korowo-rdzeniowych. Mogą mu towarzyszyć zaburzenia zwieraczowe lub czucia głębokiego.
HSP dzielimy na formy proste i złożone. W przypadku tych pierwszych niedowład jest jedynym objawem choroby. W przypadku współistniejącego zajęcia innych układów paraparezę spastyczną określamy jako złożoną. W formie złożonej HSP może dochodzić do zajęcia móżdżku, układu pozapiramidowego, nerwów obwodowych lub mięśni. W tej grupie chorych mogą występować także zaburzenia funkcji poznawczych lub psychiczne. Niekiedy u chorych występują cechy dysmorfii (zmiany w obrębie twarzy, niski wzrost), malformacje kostne (skolioza, malformacje w obrębie stóp) lub zajęcie układu wzrokowego (zaćma, zwyrodnienie barwnikowe siatkówki, zanik nerwu wzrokowego). W badaniach obrazowych chorych na HSP złożoną stwierdza się nieprawidłowości takie jak zmiany w obrębie istoty białej mózgu, ścieńczenie ciała modzelowatego, zanik rdzenia kręgowego lub móżdżku.
Klasyfikacje HSP uwzględniają sposób dziedziczenia, formę choroby (prosta lub złożona) lub zmutowany gen (SPG – spastic paraplegia genes).
Diagnostyka różnicowa jest złożona i należy wziąć pod uwagę nabyte mielopatie, ALS z dominującym uszkodzeniem górnego neuronu ruchowego oraz inne choroby o podłożu genetycznym (ataksje rdzeniowo-móżdżkowe, ataksje spastyczne) (tab. 7).
Podsumowanie
Różnicowanie choroby neuronu ruchowego powinno obejmować wiele rzadkich i trudnych do rozpoznania chorób. Pierwszym krokiem diagnostycznym powinno być ustalenie odpowiedzi na kilka kluczowych pytań, m.in. o postęp choroby, dystrybucję niedowładu, objawy towarzyszące. Pozwoli to na ustalenie wstępnego rozpoznania i zaplanowanie badań dodatkowych.
Abstract
Rare motor neuron diseases (other than amyotrophic lateral sclerosis)
A differential diagnosis of motor neuron disease should include a number of rare and difficult-to-diagnose conditions. The first diagnostic step should be to determine some key features, e.g. disease progression, distribution of paresis and associated symptoms. This helps to establish a preliminary diagnosis and plan additional examinations.
- 1. Angelini C. Genetic Neuromuscular Disorders. Springer International Publishing Switzerland 2018
- 2. Angelini C. Acquired Neuromuscular Disorders. Pathogenesis, Diagnosis and Treatment. Springer International Publishing Switzerland 2016
- 3. Bansagi B, et al. Genetic heterogeneity of motor neuropathies. Neurology 2017;88:1226-34
- 4. Beadon K, et al. Multifocal motor neuropathy. Curr Opin Neurol 2018;31:559-64
- 5. European Federation of Neurological Societies/Peripheral Nerve Society guideline on management of multifocal motor neuropathy. Report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society – first revision. J Peripher Nerv Syst 2010;15(4):295-301
- 6. Garg N, et al. Differentiating lower motor neuron syndromes. J Neurol Neurosurg Psychiatry 2017;88:474-83
- 7. Khadilkar SV, et al. Neuromuscular disorders. A comprehensive review with illustrative cases. Springer Nature Singapore Pte Ltd. 2018
- 8. Manji H, et al. Neuromuscular disease. Case studies from Queen Square. Springer-Verlag London Ltd. 2017
- 9. Rossor AM, et al. The distal hereditary motor neuropathies. J Neurol Neurosurg Psychiatry 2012;83:6e14
- 10. Sgobbi de Souza PV, et al. Hereditary Spastic Paraplegia: Clinical and Genetic Hallmarks. Cerebellum 2017;16:525-51
- 11. Statland JM, et al. Patterns of Weakness, Classification of Motor Neuron Disease & Clinical Diagnosis of Sporadic ALS. Neurol Clin 2015;33(4):735-48
- 12. Turner MR, Talbot K. Mimics and chameleons in motor neurone disease. Pract Neurol 2013;13:153-64
- 13. Walk D. Clinical Handbook of Neuromuscular Medicine. Springer International Publishing AG, part of Springer Nature 2018
- 14. Williams T. Motor neurone disease: diagnostic pitfalls. Clinical Medicine 2013;13(1):97-100


 Dodaj do ulubionych
Dodaj do ulubionych
