Nabyta hemofilia – skaza krwotoczna, o której powinien wiedzieć każdy ginekolog położnik
Jerzy Windyga
Wprowadzenie
Nabyta hemofilia A (acquired haemophilia A, AHA) jest chorobą autoimmunologiczną wywołaną przez autoprzeciwciała blokujące aktywność czynnika krzepnięcia VIII (FVIII), co prowadzi do zmniejszenia aktywności koagulacyjnej czynnika VIII (FVIII:C) w osoczu. Przeciwciała te określa się mianem inhibitora FVIII. W przeciwieństwie do wrodzonej hemofilii A, na którą chorują praktycznie wyłącznie mężczyźni, nabyta hemofilia występuje zarówno u kobiet, jak i u mężczyzn.
Występowanie i obraz kliniczny
Szacowana roczna zapadalność na nabytą hemofilię A wynosi około 1,5 na milion.1,2 Poza jednym wyjątkiem mężczyźni i kobiety są w równym stopniu narażeni na rozwój choroby. Tym wyjątkiem jest przedział wieku 20-40 lat, w którym nabyta hemofilia A jest częściej wykrywana u kobiet niż u mężczyzn.3 Wzrost zapadalności na AHA wśród młodych kobiet jest ściśle związany z okresem 6 miesięcy po porodzie.
W większości przypadków AHA objawia się nagle ciężką skazą krwotoczną, która w ciągu kilku tygodni prowadzi do zgonu około 8% osób dotkniętych chorobą.4,5 Typowe dla nabytej hemofilii A są rozległe wylewy krwi pod skórą oraz krwawienia z błon śluzowych (m.in. z dróg moczowych i rodnych), krwotoki o różnym nasileniu z ran po operacjach chirurgicznych i zabiegach ekstrakcji zębów. W odróżnieniu od klasycznej hemofilii w AHA bardzo rzadko obserwuje się samoistne wylewy krwi do stawów. Dramatyczny przebieg mają krwawienia śródczaszkowe, których zazwyczaj nie udaje się w porę zahamować. Na szczęście nie należą one do najczęstszych objawów AHA. Niebezpieczne są także krwawienia do mięśni kończyn, zwłaszcza wtedy, gdy wynaczyniona krew gromadzi się w przestrzeni anatomicznej zamkniętej powięzią. Dochodzi wówczas do ucisku na nerwy i naczynia krwionośne, którego wynikiem może być ich trwałe uszkodzenie.
U 50% chorych na AHA nie wykrywa się żadnej choroby współistniejącej (postać idiopatyczna). Około 10-15% przypadków nabytej hemofilii A dotyczy młodych kobiet w ciągu 1-6 miesięcy po porodzie, zaś w pozostałych 35-40% przypadków AHA współistnieje z chorobami autoimmunologicznymi, litymi złośliwymi guzami nowotworowymi, nowotworami hematologicznymi, chorobami alergicznymi oraz rozwija się jako odpowiedź na polekowe reakcje uczuleniowe.
Rys patofizjologiczny
Autoprzeciwciała występujące w nabytej hemofilii należą najczęściej do immunoglobulin klasy IgG. Wiążą się one z określonymi domenami (najczęściej C2 i A2) FVIII. Antykoagulacyjny mechanizm działania autoprzeciwciał polega na: 1) upośledzeniu oddziaływań FVIII z fosfolipidami (anty-C2), 2) zaburzeniach tworzenia kompleksu tenazy (anty-A2), a najprawdopodobniej także na 3) blokowaniu wiązania FVIII z czynnikiem von Willebranda.
Kinetyka oddziaływań FVIII z autoprzeciwciałami jest odmienna od obserwowanej we wrodzonej hemofilii A powikłanej alloprzeciwciałami. We wrodzonej hemofilii A alloprzeciwciała całkowicie znoszą aktywność FVIII w osoczu, natomiast w AHA, nawet przy bardzo dużym mianie autoprzeciwciał, w osoczu chorego wykrywa się resztkową aktywność FVIII. Podobnie do alloprzeciwciał autoprzeciwciała wobec FVIII nie wiążą dopełniacza i nie powodują reakcji alergicznych.
Szczególna postać AHA – nabyta hemofilia A po porodzie
Nabyta hemofilia A jest stosunkowo rzadkim powikłaniem ciąży i połogu, gdyż ocenia się, że występuje nie częściej niż w jednym na 350 000 porodów.2,6 Choroba może wystąpić natychmiast po porodzie, objawiając się ciężkim krwotokiem z dróg rodnych lub do 6 miesięcy po urodzeniu dziecka i wówczas dominującym objawem są podskórne krwiaki, niekiedy obejmujące znaczną powierzchnię ciała, którym mogą towarzyszyć mniej lub bardziej nasilone krwotoki śluzówkowe. Dane rejestru European Acquired Hemophilia Registry (EACH2) wskazują, że nabytą hemofilię związaną z ciążą i połogiem najczęściej wykrywa się w ciągu 25-180 dni po porodzie (mediana 89 dni), a skaza krwotoczna objawia się najczęściej samoistnymi krwiakami śródmięśniowymi oraz krwawieniami zaotrzewnowymi.7
W około 70% przypadków AHA związanej z ciążą/połogiem można spodziewać się samoistnej remisji choroby w ciągu 30 dni od rozpoznania. Jeśli jednak do remisji nie dochodzi, a skaza krwotoczna ma ciężki przebieg, należy włączyć odpowiednie leczenie hemostatyczne i immunosupresyjne (patrz niżej). W opinii wielu klinicystów eradykacja inhibitora FVIII za pomocą leków immunosupresyjnych u kobiety z AHA po porodzie jest często trudniejsza niż w idiopatycznej postaci choroby bądź w AHA na podłożu innych chorób i stanów klinicznych.2 Opinia ta stoi w sprzeczności z wynikami rejestru EACH2, z których wynika, że glikokortykosteroidy skuteczniej eliminują inhibitor FVIII u kobiet z AHA po porodzie w porównaniu do innych podgrup chorych na AHA.7 Inhibitor FVIII związany z ciążą i połogiem częściej pojawia się u pierwiastek i raczej nie wykazuje tendencji do nawrotów w kolejnych ciążach. W jednej pracy opisano zmniejszenie aktywności FVIII we krwi noworodka urodzonego przez kobietę z AHA.8
Diagnostyka laboratoryjna
Typowa konstelacja wyników przesiewowych badań hemostazy u chorego na AHA to: wydłużenie (najczęściej 2-3-krotne) czasu częściowej tromboplastyny po aktywacji (activated partial thromboplastin time, APTT), przy prawidłowych wartościach czasów protrombinowego (prothrombin time, PT), trombinowego (thrombin time, TT), krwawienia (bleeding time, BT) oraz przy prawidłowej liczbie płytek krwi i prawidłowym stężeniu fibrynogenu. Taki układ wyników badań występuje jeszcze tylko we wrodzonych niedoborach czynników krzepnięcia VIII, IX, XI i XII oraz w przypadku obecności w badanym osoczu antykoagulantu toczniowego, który – jak wiadomo – in vivo nie wywołuje skłonności do krwawień, ale predysponuje do zakrzepów. Jeśli przyczyną wydłużonego APTT jest obecność w próbce krwi heparyny niefrakcjonowanej, czas trombinowy jest nieoznaczalny.
W celu potwierdzenia rozpoznania AHA przeprowadza się tzw. test korekcji APTT, który polega na pomiarze APTT w mieszaninie równych objętości osocza badanego i prawidłowego. U osoby z AHA APTT takiej mieszaniny nie skraca się istotnie (inhibitor neutralizuje FVIII obecny w próbce prawidłowego osocza). Następnie oznacza się aktywność FVIII, która u zdrowych osób mieści się w przedziale 50-150 j.m./dl (50-150% normy), zaś u chorych na AHA wynosi zwykle 0-15 j.m./dl (0-15% normy). Ostatnim etapem laboratoryjnej diagnostyki inhibitora FVIII jest oznaczenie jego miana, wyrażonego w jednostkach Bethesda (j.B./ml).
Leczenie
Strategia postępowania z chorym na AHA obejmuje dwa główne cele: 1) doraźny, którym jest leczenie i profilaktyka krwawień oraz 2) długofalowy, polegający na eliminacji inhibitora (rycina).
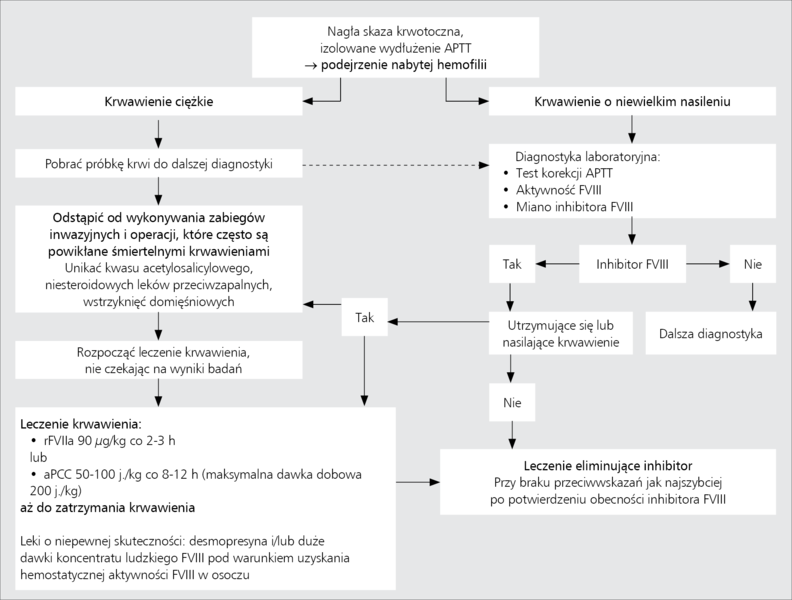
Rycina. Algorytm postępowania z chorym na nabytą hemofilię [za poz. 5 w modyfikacji].
Najlepszym sposobem leczenia krwawień w przebiegu AHA jest stosowanie rekombinowanego aktywowanego czynnika VII (recombinant activated factor VII, rFVIIa) lub koncentratu aktywowanych czynników zespołu protrombiny (activated prothrombin complex concentrate, aPCC) (tabela). Koncentraty te aktywują krzepnięcie krwi z pominięciem etapu zależnego od FVIII. Skuteczność rFVIIa i aPCC w leczeniu krwawień w przebiegu nabytej hemofilii została potwierdzona wynikami badań klinicznych.9,10 Największą wadą obu leków jest brak możliwości laboratoryjnej kontroli ich skuteczności. Należy także pamiętać o ryzyku powikłań zakrzepowych, na jakie potencjalnie narażają oba leki, szczególnie u osób starszych ze współistniejącymi czynnikami ryzyka żylnej choroby zakrzepowo-zatorowej czy zakrzepicy tętniczej, a także, choć w mniejszym stopniu, u kobiet w połogu. W wyjątkowych przypadkach AHA o małym mianie inhibitora wobec FVIII i krwawień o niewielkim nasileniu skuteczne może się okazać zastosowanie koncentratu ludzkiego FVIII lub desmopresyny (tabela).