ŚWIĄTECZNA DARMOWA DOSTAWA od 20 grudnia do 8 stycznia! Zamówienia złożone w tym okresie wyślemy od 2 stycznia 2025. Sprawdź >
Specjalista-pediatrze
RADA NAUKOWA DZIAŁU dr n. med.Anna Zawadzka-Krajewska, dr n. med. Małgorzata Bartkowiak-Emeryk, dr hab. n. med. Jerzy Ziołkowski
Zespół hiper-IgE (HIES) – implikacje kliniczne
Katarzyna Grzela
Pediatr Dypl. 2012;16(4):64-67
Wstęp
Zespół hiper-IgE (hyper-IgE syndrome, HIES), w piśmiennictwie opisywany również pod nazwą zespołu Joba (Hioba), jest rzadką chorobą o charakterze pierwotnego niedoboru odporności. Charakterystyczna dla HIES triada objawów – nawracające zakażenia, zmiany skórne oraz zwiększone stężenie immunoglobulin klasy E (IgE) w surowicy – pojawia się jednak również w innych jednostkach chorobowych.
Nawracające zakażenia, zwłaszcza dróg oddechowych, z towarzyszącymi im zmianami skórnymi, często występują u dzieci, stanowiąc istotny problem w codziennej praktyce klinicznej. Jeśli w wykonywanych w ramach diagnostyki badaniach laboratoryjnych stwierdza się duże stężenia IgE, wówczas u większości takich chorych rozpoznaje się atopię. Warto jednak pamiętać, że duże stężenie IgE, zwykle ze znaczną eozynofilią, obok infekcji pasożytniczych może towarzyszyć również wspomnianemu zespołowi hiper-IgE. W diagnostyce różnicowej należy uwzględnić także m.in. aspergilozę płucną, szpiczaka wytwarzającego IgE oraz inne rzadziej występujące zespoły – Nethertona, Wiskotta-Aldricha czy Ommena.
W przypadku niedoboru odporności, jakim jest HIES, możliwie wczesne wdrożenie odpowiedniej profilaktyki przeciwbakteryjnej jest istotne dla zapobiegania wystąpieniu ewentualnych ciężkich powikłań po zakażeniu. Ze względu na różny sposób dalszego postępowania z chorym sama świadomość możliwości występowania zespołu hiper-IgE, a tym bardziej umiejętność odróżnienia go od alergii atopowej, stają się bardzo ważne dla lekarza pediatry i jeszcze ważniejsze dla przyszłych losów dziecka. Dlatego też celem niniejszego opracowania jest przybliżenie tej rzadkiej jednostki chorobowej.
Epidemiologia
Zespół hiper-IgE występuje z częstością ok. 1:500 000-1:1 000 000 przypadków. Dotychczas nie wykazano wyraźnych predyspozycji do jego rozwoju ze względu na płeć ani rasę, zwraca jednak uwagę występowanie rodzinne, sugerujące genetyczne podłoże tej choroby. Analiza sposobu dziedziczenia oraz badania molekularne potwierdziły te przypuszczenia, pozwalając jednocześnie na wyróżnienie dwóch postaci zespołu – częściej spotykanej, dziedziczonej autosomalnie dominująco (AD-HIES), oraz nieco rzadszej – dziedziczonej autosomalnie recesywnie (AR-HIES). Początkowo uważane za tę samą patologię o zróżnicowanym nasileniu objawów, dzięki identyfikacji mutacji w odrębnych genach obecnie obie formy – AD-HIES i AR-HIES – choć podobne, uznawane są za odrębne jednostki chorobowe.
Objawy
Objawy kliniczne HIES pojawiają się zwykle bardzo wcześnie. Pierwsze zmiany skórne, często identyfikowane jako atopowe, zlokalizowane są na skórze twarzy i głowy. W odróżnieniu od atopowego zapalenia skóry zmiany te pojawiają się jednak już w pierwszych dobach życia, nie towarzyszą im inne objawy atopii. W badaniach laboratoryjnych stwierdza się stężenia IgE w surowicy znacznie przekraczające 2000 IU/ml, zwykle powyżej 10-krotności górnego zakresu normy (lub 95 centyla), oraz eozynofilię, zazwyczaj powyżej 700 komórek w μl.1
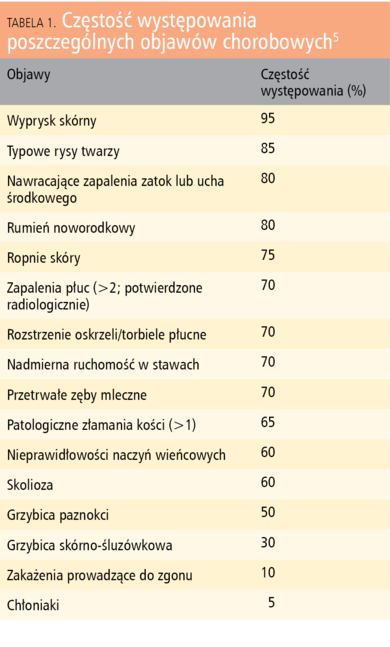
Tabela 1. Częstość występowania poszczególnych objawów chorobowych5
Patognomoniczne dla HIES są głębokie ropnie skórne o etiologii gronkowcowej, nazywane ropniami zimnymi. Badanie histopatologiczne biopsji skóry wykazuje nacieki eozynofilowe. Inne charakterystyczne zmiany skórne to: kandydoza skórno-śluzówkowa wywołana przez Candida albicans, grzybica paznokci, zdarzają się również miejscowe powikłania poszczepienne, zwłaszcza po podaniu BCG.2
Ze strony układu oddechowego typowe dla zespołu hiper-IgE są przede wszystkim nawracające zapalenia płuc, głównie o etiologii gronkowcowej (Staphylococcus aureus), w tym również szczepami metycylinoopornymi (MRSA), ale także wywołane bakteriami otoczkowymi – Streptococcus pneumoniae i Haemophilus influenzae.3 Często, jako powikłanie zapalenia płuc, pojawiają się ropnie płuc, tworzą się rozstrzenia oskrzeli i przetoki oskrzelowo-płucne. Niekorzystnym następstwem zapalenia płuc w postaci AD-HIES, niespotykanym w AR-HIES, może być wytworzenie torbieli płucnych (pneumatocele), kolonizowanych przez pałeczki ropy błękitnej (Pseudomonas aeruginosa) czy kropidlaki Aspergillus fumigatus. Opisywane były również przypadki krwioplucia prowadzącego do zgonu w przebiegu zakażenia Ps. aeruginosa, Aspergillus fumigatus lub Scedosporium prolificans.4 Zmiany zapalne mogą obejmować również górne drogi oddechowe, gdzie zwykle przyjmują postać przewlekłego zapalenia zatok przynosowych, obserwowano także wysiękowe zapalenie ucha środkowego oraz zakażenia wewnątrzmózgowe.4
Obok wymienionych powyżej objawów klinicznych (z wyjątkiem tworzenia torbieli płucnych), wspólnych dla obu postaci HIES, AD-HIES cechuje dodatkowo występowanie charakterystycznych, „grubych” rysów twarzy: wystające czoło, głęboko osadzone oczy, szeroka nasada nosa i pogrubiała dolna warga. Towarzyszą im nieprawidłowości rozwoju zębów – opóźnione wypadanie zębów mlecznych, zaburzenia w budowie szkliwa, nasilona próchnica, obecność gotyckiego podniebienia, jak również patologie dotyczące tkanki kostnej – nieadekwatna do siły urazu skłonność do złamań kości długich, żeber i, nieco rzadziej, kręgosłupa. U ok. 60% dzieci z AD-HIES występuje różnie nasilona skolioza. Inne zaburzenia tkanki łącznej to m.in. nadmierna wiotkość stawów, tętniaki aorty piersiowej, malformacje naczyniowe, jak również zmiany o charakterze autoimmunologicznym – toczeń rumieniowaty układowy (SLE) czy zapalenie skórno-mięśniowe (dermatomyositis). Obserwuje się również skłonność do nowotworzenia, szczególnie do powstawania chłoniaków zarówno nieziarniczych, jak i ziarnicy złośliwej (choroby Hodgkina).5
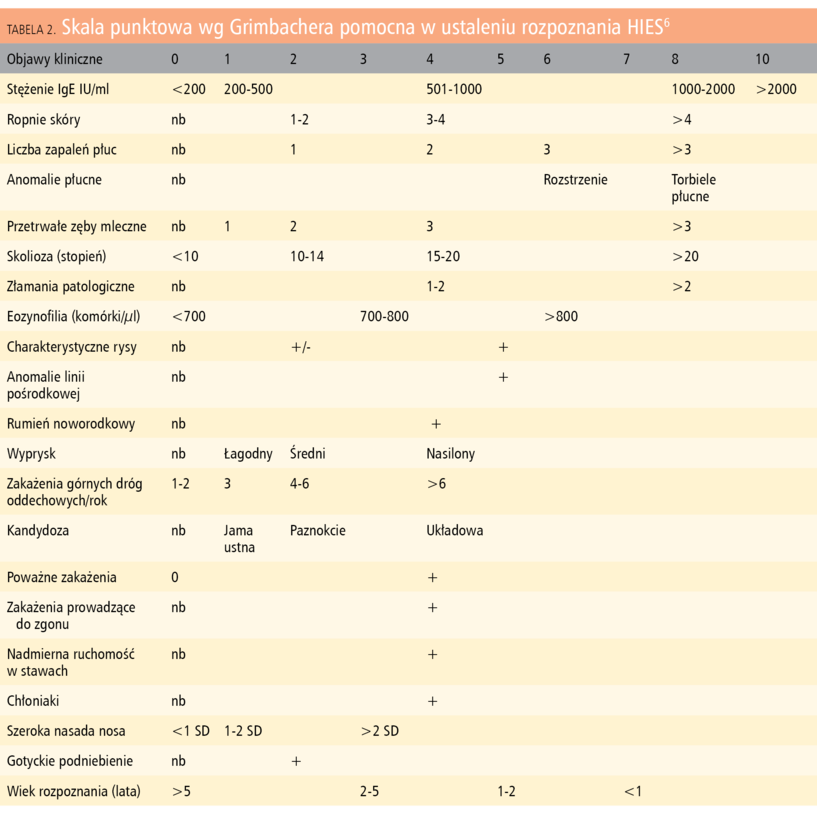
Tabela 2. Skala punktowa wg Grimbachera pomocna w ustaleniu rozpoznania HIES6
Obecność charakterystycznych objawów oraz duże stężenie IgE w surowicy stanowią podstawę rozpoznania HIES. W ustaleniu rozpoznania pomocna jest skala punktowa (wg Grimbachera6), przedstawiona w tabeli 2.
Otrzymanie wyniku <15 punktów we wspomnianej skali pozwala z dużym prawdopodobieństwem wykluczyć obecności HIES. Wyniki w przedziałach 16-39 punktów wskazują na możliwość, a 40-59 punktów – na duże prawdopodobieństwo choroby. Wynik powyżej 60 oznacza, że chory na pewno jest nosicielem zmutowanego genu. Chorzy uzyskujący wysokie punktacje powinni być kierowani do ośrodków wysokospecjalistycznych w celu weryfikacji rozpoznania metodami molekularnymi.
Podłoże molekularne HIES
Analiza próbek DNA chorych z objawami zespołu hiper-IgE oraz ich krewnych pozwoliła na wyróżnienie wspomnianych wcześniej dwóch genetycznych odmian zespołu – AR-HIES i AD-HIES. Wariant dziedziczony autosomalnie recesywnie dotyczy dzieci spokrewnionych rodziców i związany jest z mutacją genu dla kinazy tyrozynowej 2 (TYK2). Produkt genu, kinaza Tyk2, uczestniczy w przekazywaniu do wnętrza komórek sygnału z receptorów dla różnych cytokin, m.in. interferonów, interleukiny (IL)-12, IL-23, a pośrednio również IL-6 i IL-10. Z tego powodu mutacja genu TYK2 powoduje poważne zaburzenia immunologiczne, w tym znaczne upośledzenie odporności przeciwwirusowej.7
U większości chorych z wariantem autosomalnym dominującym zidentyfikowano w sumie 5 dominujących negatywnych mutacji genu dla regulatora transkrypcji STAT3. Badając 50 chorych na HIES i 48 członków ich rodzin stwierdzono, że transmisja rodzinna mutacji występowała u blisko 34%, aż 52% stanowiły mutacje sporadyczne, natomiast 14% przypadków dotyczyło mutacji powstałych de novo.