


Co znajdziesz w artykule?
- Obraz kliniczny zespołu Turnera – korelacja pomiędzy kariotypem a fenotypem
- Strategia leczenia niedoboru wzrostu będącego głównym objawem zespołu Turnera
- Opieka nad pacjentkami z zespołem Turnera
Spis treści
Zespół Turnera (ZT) zaliczany jest do chorób rzadkich. Częstość jego występowania szacuje się na 25-50 przypadków/100 000 żywo urodzonych noworodków płci żeńskiej, co biorąc pod uwagę liczbę urodzeń w Polsce, przekłada się na 80-100 nowych rozpoznań rocznie (ICD-10: Q96). ZT jest konsekwencją całkowitej lub częściowej monosomii chromosomu X obecnej we wszystkich bądź tylko w niektórych liniach komórkowych. Diagnozę ZT można postawić jedynie u dziewczynki/kobiety z żeńskim fenotypem (żeńskimi
narządami płciowymi), u której jednocześnie stwierdza się cechy zespołu (tab. 1) współistniejące z charakterystycznym kariotypem w badaniu cytogenetycznym 1 (tab. 2).
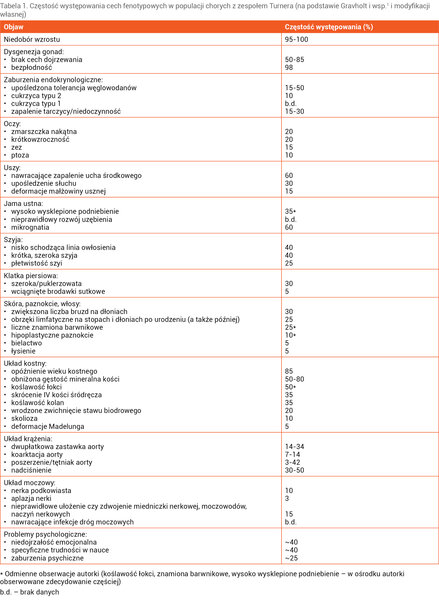
Tabela 1. Częstość występowania cech fenotypowych w populacji chorych z zespołem Turnera (na podstawie Gravholt i wsp.1 i modyfikacji własnej)
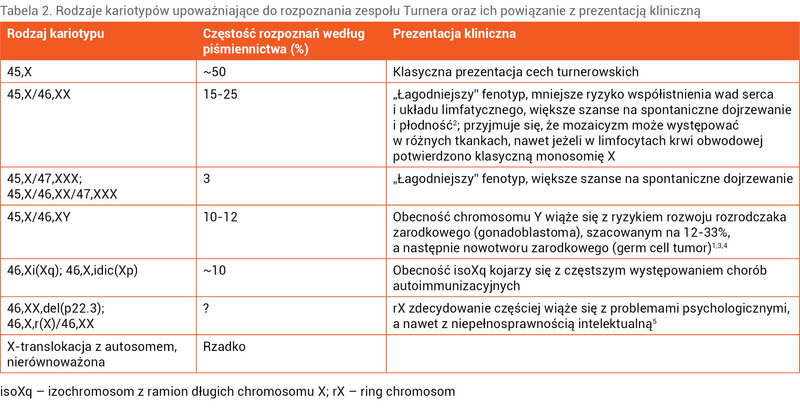
Tabela 2. Rodzaje kariotypów upoważniające do rozpoznania zespołu Turnera oraz ich powiązanie z prezentacją kliniczną
Badanie cytogenetyczne potwierdzające rozpoznanie ZT
Zgodnie z zaleceniami American College of Medical Genetics badanie kariotypu powinno zostać wykonane na podstawie analizy co najmniej 20 komórek 6 . W przypadku rozpoznań/podejrzeń prenatalnych kariotyp należy ocenić po urodzeniu dziecka. Z uwagi na znaczny postęp w technikach badania sugeruje się również powtórzenie oznaczenia kariotypu u tych pacjentek, u których badanie to wykonano wiele lat temu 1 .
Nie każda częściowa utrata materiału genetycznego jednego z chromosomów X pozwala na rozpoznanie ZT. W przypadku delecji niewielkiej części ramienia długiego dystalnie do Xq24 możemy spodziewać się izolowanego pierwotnego lub wtórnego braku miesiączki, co nie daje podstawy do rozpoznania ZT. U około 50% pacjentek z ZT w klasycznym badaniu cytogenetycznym z limfocytów krwi obwodowej potwierdza się monosomię 45,X. U prawie 1/3 stwierdza się kariotyp mozaikowy z obecną prawidłową linią 46,XX. U pozostałych występują inne anomalie strukturalne 1 chromosomu X (tab. 2).
Prawidłowy wynik badania cytogenetycznego z limfocytów krwi obwodowej u pacjentki z cechami turnerowskimi powinien skłaniać do poszerzenia diagnostyki o kariotyp z fibroblastów skóry 1 .
Choć nie potwierdzono ścisłej korelacji genotyp–fenotyp, to niektóre rodzaje kariotypów mogą sugerować specyficzny przebieg kliniczny ZT i większe ryzyko chorób współistniejących (tab. 2).
Fenotyp pacjentki z ZT
Rozpoznanie ZT może zostać ustalone przez różnych specjalistów, co wynika z wielu współistniejących problemów zdrowotnych, oraz na różnym etapie życia pacjentki, co z kolei jest związane z różnym nasileniem fenotypu (tab. 1). Wystąpienie cech fenotypowych (tzw. stygmatów), które składają się na zespół objawów ZT, tłumaczy się haploinsuficjencją genu/genów (obecnością tylko jednej prawidłowej kopii genu/genów) zlokalizowanych na chromosomie X 7 . W prenatalnym badaniu ultrasonograficznym podejrzenie ZT budzą: obrzęk płodu, wodniak torbielowaty, wada układu krążenia czy układu moczowego. Nieprawidłowe stężenia w surowicy α-fetoproteiny, gonadotropiny kosmówkowej, inhibiny A czy wolnego estriolu (test potrójny, test poczwórny) również mogą nasuwać podejrzenie ZT 1 . Po urodzeniu się dziecka uwagę zwracają: mała urodzeniowa masa i długość ciała, obrzęki limfatyczne stóp i dłoni, płetwistość szyi, deformacja małżowin usznych oraz ich niskie osadzenie, wysoko wysklepione podniebienie oraz zmarszczka nakątna.
W przypadku obecności opisanych cech fenotypowych u noworodka płci żeńskiej konieczne jest wykluczenie typowych wad układu krążenia, w tym przede wszystkim dwupłatkowej zastawki aorty (BAV – bicuspid aortic valve) oraz koarktacji aorty (CoA – coarctation of the aorta), występujących odpowiednio u 16% i 11% pacjentek z ZT. Zasady postępowania w przypadku stwierdzenia wad układu sercowo-naczyniowego zostały bardzo precyzyjnie przedstawione w ostatnio opublikowanych zaleceniach. Poza klasyczną konsultacją kardiologiczną z badaniem elektrokardiograficznym oraz echokardiograficznym, monitorowaniem ciśnienia tętniczego konieczne jest wykonanie rezonansu magnetycznego (MR) w celu pomiaru aorty i oszacowania ryzyka wystąpienia tętniaka rozwarstwiającego aorty. Jest to szczególnie istotne w przypadku młodych kobiet z ZT, u których pęknięcie tętniaka jest najczęstszą przyczyną nagłego zgonu (ryzyko pęknięcia tętniaka znacznie wzrasta w okresie ciąży – spontanicznej lub po zastosowaniu technik wspomaganego rozrodu). Problem nadciśnienia tętniczego narasta z wiekiem (u blisko 30% dziewcząt i 50% dorosłych z ZT) i podobnie jak w przypadku CoA oraz BAV istotnie zwiększa ryzyko rozwarstwienia aorty. Współistniejące wady układu moczowego (nerka podkowiasta czy zdwojenie układu kielichowo-miedniczkowego) mogą przyczyniać się do nawracających zakażeń w drogach moczowych oraz wystąpienia lub nasilenia nadciśnienia tętniczego.
Od okresu przedszkolnego u chorych z ZT charakterystyczne jest zwolnienie tempa wzrastania i niedobór wzrostu. U nastolatek uwagę zwracają brak cech dojrzewania płciowego lub opóźnione dojrzewanie czy zaburzenia miesiączkowania, często pod postacią pierwotnego lub wtórnego braku miesiączki 2 . ZT predysponuje do dysgenezji gonad i jest klasycznym przykładem hipogonadyzmu hipergonadotropowego. Ponad 90% dziewcząt i kobiet wymaga hormonalnej terapii zastępczej. Gonadotropiny monitoruje się od 10-11 roku życia. Ich wysoka wartość (stężenie hormonu folikulotropowego (FSH – follicle-stimulating hormone) >10 j.m./ml) przy braku rozwoju gruczołów piersiowych może wskazywać na potrzebę indukcji dojrzewania (11-12 rok życia) z zastosowaniem bardzo małych dawek naturalnych estrogenów. W celu zoptymalizowania terapii proponuje się estradiol w formie transdermalnej lub doustnej w dawkach początkowych stanowiących 1/8-1/10 dawki docelowej, do której dochodzi się stopniowo przez 2-3 lata. Dołączenie w odpowiednim momencie progestagenów jest konieczne, aby zapobiec przerostom endometrium 1, 8 .
ZT często kojarzy się ze zwiększonym ryzykiem wystąpienia chorób autoimmunizacyjnych, stąd zarówno w chwili rozpoznania zespołu, jak i w trakcie obserwacji konieczne jest wykonanie badań i monitorowanie w kierunku: choroby Hashimoto (autoimmunizacyjnego zapalenia tarczycy), celiakii, cukrzycy, bielactwa, łysienia plackowatego czy nieswoistego zapalenia jelit 9, 10 . Częstym i narastającym z wiekiem problemem są zaburzenia słuchu, w dzieciństwie występujące głównie wskutek nawracających zapaleń ucha środkowego (niedosłuch przewodzeniowy), natomiast w życiu dorosłym mające charakter niedosłuchu odbiorczego.
Diagnoza ZT zobowiązuje do przeprowadzania u chorych regularnych badań kontrolnych, w dużej mierze wykonywanych cyklicznie jako badania profilaktyczne, oraz w przypadku potwierdzenia chorób współistniejących wymaga objęcia pacjentek stałą opieką wielospecjalistyczną (tab. 3).
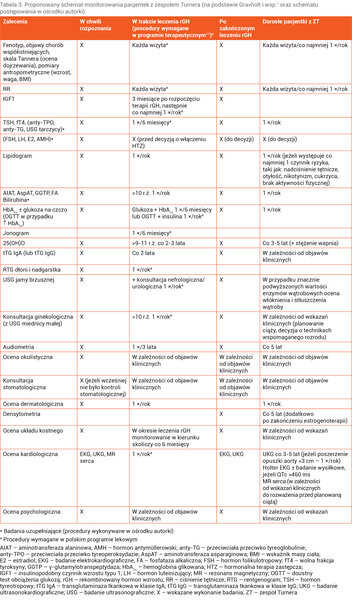
Tabela 3. Proponowany schemat monitorowania pacjentek z zespołem Turnera (na podstawie Gravholt i wsp.1 oraz schematu postępowania w ośrodku autorki)
Stopień nasilenia dysmorfii może być różny i niekoniecznie wpływa na szybkość postawienia diagnozy. Średni wiek rozpoznania ZT w populacji polskiej szacuje się na 9-10 rok życia, czyli 2 lata później niż w populacji amerykańskiej 12, 13 . Najczęstszymi powodami zlecenia wykonania badania cytogenetycznego są niedobór wzrostu stwierdzany u prawie wszystkich pacjentek oraz objawy związane z niewydolnością gonad obserwowane w różnym nasileniu u około 90% zdiagnozowanych.
Spontaniczne wzrastanie w ZT
Niedobór wzrostu jest głównym objawem ZT i dotyczy 95-100% badanych 1 . Ich spontaniczne wzrastanie w ZT ma charakterystyczny przebieg i w ciągu życia wyróżnia się kilka jego faz:
- okres wewnątrzmacicznego zahamowania wzrastania (IUGR – intrauterine growth restriction)
- okres od urodzenia do 3 roku życia z prawidłowym tempem wzrastania i spowolnioną progresją wieku kostnego w stosunku do wieku metrykalnego
- okres od 3 do 12 roku życia z opóźnionym tempem wzrastania i prawidłową progresją wieku kostnego
- okres po 12 roku życia z opóźnionym tempem wzrastania i spowolnioną progresją wieku kostnego.
Konsekwencją opóźnienia wieku kostnego jest wydłużenie okresu wzrastania nawet do początku trzeciej dekady życia. Nie wiąże się to jednak z poprawą prognozy wzrostu ostatecznego. Wzrost końcowy nieleczonych pacjentek jest przeciętnie o 18-20 cm niższy od prognozowanego (ryc. 1). W celu wiarygodnej oceny spontanicznego tempa wzrastania zaleca się używanie siatek centylowych dla ZT, a w przypadku stwierdzenia odchylania się od dotychczasowego kanału centylowego ważne jest poszukiwanie dodatkowej przyczyny nieprawidłowego wzrastania (np. niedoczynności tarczycy w przebiegu przewlekłego zapalenia, celiakii czy nieswoistego zapalenia jelit).
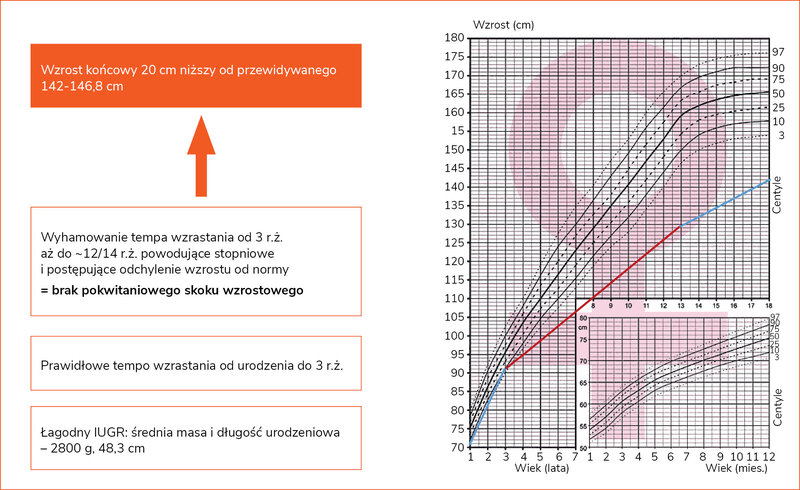
Rycina 1. Przykładowy spontaniczny przebieg tempa wzrastania u nieleczonej pacjentki z zespołem Turnera
Niskorosłość w ZT nie wynika z niedoboru hormonu wzrostu. Za zwolnienie tempa wzrastania oraz niski wzrost w dużej mierze odpowiada haploidalność genu SHOX (short stature homeobox-containing gene) zlokalizowanego w pseudoautosomalnym regionie ramienia krótkiego chromosomów płciowych. Gen ten koduje czynnik transkrypcyjny, kluczowy dla prawidłowego rozwoju układu kostnego, w szczególności kości kończyn.
Terapia hormonalna poprawiająca wzrost końcowy
W Polsce ZT jest jednym ze wskazań do refundacji leczenia hormonem wzrostu. Historia stosowania hormonu wzrostu w terapii niedoboru wzrostu sięga lat 60. XX wieku, a jego skuteczność w populacji z ZT potwierdzono w opublikowanym w 2005 roku badaniu kanadyjskim (ryc. 2), które było badaniem randomizowanym kontynuowanym do osiągnięcia niemalże wzrostu ostatecznego 14 . Pacjentki były leczone od 5,5-7,6 roku życia i w porównaniu z obserwowaną w tym czasie grupą kontrolną ich wzrost końcowy poprawił się o 5-8 cm. Podobne efekty obserwowano też w innych badaniach 15 .

Rycina 2. Rys historyczny wykorzystania hormonu wzrostu w terapii niskorosłości u pacjentek z zespołem Turnera
Uwzględniając profil spontanicznego wzrastania (ryc. 1), leczenie rekombinowanym hormonem wzrostu (rGH – recombinant growth hormone) dziewcząt z ZT optymalnie należałoby rozpoczynać w 4-6 roku życia, aby uzyskać wzrost w granicach normy wiekowej i uniknąć dużych różnic w rozwoju fizycznym w porównaniu z rówieśniczkami. Oczywiście czas rozpoczęcia terapii w dużej mierze zależy od wieku, w którym postawiono diagnozę. Dla uzyskania oczekiwanych korzyści z leczenia powinno ono zostać zainicjowane przed 12-13 rokiem życia. W polskich warunkach refundacja leczenia w ramach programu terapeutycznego przysługuje tylko w przypadku wzrostu poniżej 3 pc (percentyla) według siatek centylowych dla zdrowej populacji żeńskiej. Międzynarodowe towarzystwa rekomendują rozpoczęcie terapii także w przypadku zwolnienia tempa wzrastania (<50 pc na podstawie 6-miesięcznej obserwacji), jak również w przypadku niekorzystnej prognozy wzrostowej wynikającej np. z niskiego wzrostu rodziców czy zaawansowanego dojrzewania (dojrzewające spontanicznie dziewczęta z ZT mają bardziej zaawansowany wiek kostny, co skraca okres wzrastania i ostatecznie skazuje na niski wzrost końcowy) 1 .
Leczenie rGH rozpoczyna się od dawki 45-50 µg/kg/24 h (odpowiednik około 1 j.m./kg/tydzień lub 1,3-1,5 mg/m 2 / 24 h bądź 4-4,5 j.m./m 2 /24 h) stosowanej w codziennych wieczornych podskórnych iniekcjach. Szkolenie rodziców/opiekunów oraz pacjentek w zakresie obsługi penów do podawania leku w warunkach domowych odbywa się w ośrodkach prowadzących programy terapeutyczne. Dawka rGH jest systematycznie modyfikowana przez lekarza prowadzącego podczas wizyt kontrolnych odbywających się najczęściej co 3-6 miesięcy. Poza masą/powierzchnią ciała bierze się pod uwagę efekty terapeutyczne (tempo wzrastania) oraz stężenie insulinopodobnego czynnika typu 1 (IGF1 – insulin-like growth factor type 1). W większości krajów kryterium zakończenia terapii rGH jest wiek kostny 14 lat oraz wyhamowanie wzrastania poniżej 2-3 cm w skali roku, a według standardów polskich również osiągnięcie wysokości 158 cm (10 pc według siatek dla zdrowej populacji).
Czynnikami predykcji wyższego wzrostu końcowego po zakończeniu terapii rGH są:
- wyższy wzrost w chwili inicjacji leczenia
- wyższy wzrost rodziców (większa średnia wysokość ciała rodziców [MPH – mid-parental height])
- młodszy wiek rozpoczęcia leczenia
- dłuższy czas stosowania rGH przed rozpoczęciem dojrzewania
- dłuższy czas leczenia i większa dawka rGH 1 .
Za wiarygodny wskaźnik odpowiedzi na leczenie przyjmuje się tempo wzrastania w pierwszych 2 latach terapii. Znaczenie mają też kontrolowanie i leczenie chorób towarzyszących – w każdym przypadku niewystarczającej odpowiedzi na rGH należy wykluczyć współistnienie chorób przewlekłych (np. zaburzeń wchłaniania w przebiegu celiakii, niedoboru witaminy D3, niedoczynności tarczycy).
Przed rozpoczęciem terapii należy poinformować o potencjalnie mogących wystąpić działaniach niepożądanych. Włączenie leczenia nakłada obowiązek ścisłego monitorowania gospodarki węglowodanowej, zwrócenia uwagi na objawy wzmożonego ciśnienia śródczaszkowego oraz złuszczenia głowy kości udowej 16 , obserwacji w kierunku rozwoju skoliozy bądź pogłębiania się już istniejącej 17 , objawów zapalenia trzustki 1 . W trakcie leczenia należy unikać zbyt dużych stężeń IGF1 (oznaczenia kontrolne wykonywane są przynajmniej raz w roku), tj. powyżej 2 odchyleń standardowych (SD – standard deviation). W przypadku podwyższonych stężeń IGF1 wskazana jest redukcja dawki rGH. Dane z dużego rejestru nie potwierdziły związku stosowania rGH ze zwiększonym ryzykiem rozrostu nowotworowego w populacji ZT 17, 18, 19 .
W przypadku znacznego deficytu wzrostowego i słabej prognozy wzrostu końcowego u dziewcząt powyżej 10 roku życia można rozważyć dołączenie steroidu anabolicznego (oksandrolonu; w Polsce sprowadzany w ramach importu docelowego). Większe dawki tego leku mogą niekorzystnie wpływać na rozwój gruczołów piersiowych, być przyczyną objawów wirylizacji (powiększenie łechtaczki, niski głos, trądzik, hirsutyzm) oraz przyspieszenia wieku kostnego, dlatego rekomenduje się podawanie niższych dawek – 0,03 mg/kg/24 h. W trakcie leczenia wskazane jest monitorowanie enzymów wątrobowych. Korzyść wynikająca z zastosowania tego leku w terapii skojarzonej z rGH to wzrost końcowy wyższy o 2-5 cm w porównaniu z osobami leczonymi jedynie rGH 1, 20, 21 . Dołączanie bardzo małych dawek estrogenów do leczenia rGH przed okresem fizjologicznego dojrzewania pacjentki w celu poprawy wzrostu nie jest aktualnie rekomendowane i wymaga dalszych badań 1 .
Artykuł powstał we współpracy z Sandoz Polska Sp. z o.o. SPEAK/MED/61/06-2020
Abstract
Diagnostic and therapeutic management of Turner syndrome
Turner syndrome is a genetic syndrome in girls associated with complete or partial monosomy of chromosome X that is present in all or only some cell lines. The consequences include a short stature and abnormalities in the course of sexual maturation. It is estimated that 80-100 neonates are diagnosed with Turner syndrome in Poland annually. The wide range of health problems encountered in individuals with Turner syndrome means that they need to have access to multispeciality medical care.
This article describes the clinical presentation of Turner syndrome and discusses the phenotype-karyotype correlation. A short stature is seen in 95-100% of patients with Turner syndrome, making it the most prominent manifestation. Accordingly, much space in the article is devoted to the strategy for treating short stature, and particularly the use of recombinant growth hormone in hormone therapy to improve adult body height. The author also proposes a scheme for monitoring patients with Turner syndrome on the basis of the relevant literature and her own experience.
- 1. Gravholt CH, Andersen NH, Conway GS, et al.; International Turner Syndrome Consensus Group. Clinical practice guidelines for the care of girls and women with Turner syndrome: proceedings from the 2016 Cincinnati International Turner Syndrome Meeting. Eur J Endocrinol 2017;177(3):G1-70
- 2. Hankus M, Sołtysik K, Szeliga K, et al. Prediction of spontaneous puberty in Turner syndrome based on mid-childhood gonadotropin concentrations, karyotype and ovary visualization: a longitudinal study. Horm Res Paediatr 2018;89(2):90-7
- 3. Mazzanti L, Cicognani A, Baldazzi L, et al. Gonadoblastoma in Turner syndrome and Y-chromosome-derived material. Am J Med Genet 2005;135:150-4
- 4. Cools M, Drop SL, Wolffenbuttel KP, et al. Germ cell tumors in the intersex gonad: old paths, new directions, moving frontiers. Endocr Rev 2006;27:468-84
- 5. Leppig KA, Sybert VP, Ross JL, et al. Phenotype and X inactivation in 45,X/46,X,r(X) cases. Am J Med Genet A 2004;128A(3):276-84
- 6. Wolff DJ, Van Dyke DL, Powell CM; Working Group of the ACMG Laboratory Quality Assurance Committee. Laboratory guideline for Turner syndrome. Genet Med 2010;12(1): 52-5
- 7. Davenport ML. Approach to the patient with Turner syndrome. J Clin Endocrinol Metab 2010;95:1487-95
- 8. Donaldson M, Kriström B, Ankarberg-Lindgren C, et al.; on behalf of the European Society for Paediatric Endocrinology Turner Syndrome Working Group. Optimal pubertal induction in girls with Turner syndrome using either oral or transdermal estradiol: a proposed modern strategy. Horm Res Paediatr 2019;91(3):153-63
- 9. Węgiel M, Antosz A, Gieburowska J, et al. Autoimmunity predisposition in girls with Turner syndrome. Front Endocrinol (Lausanne) 2019;10:511
- 10. Gawlik AM, Berdej-Szczot E, Blat D, et al. Immunological profile and predisposition to autoimmunity in girls with Turner syndrome. Front Endocrinol (Lausanne) 2018;9:307
- 11. Ministerstwo Zdrowia. Załącznik B.42. Leczenie niskorosłych dzieci z zespołem Turnera. https://www.gov.pl/attachment/24fb030d-07ac-44a5-afbb-866296323384. Dostęp 5.10.2020
- 12. Gawlik A, Gawlik T, Małecka-Tendera E i wsp. Wpływ nasilenia fenotypu na czas rozpoznawania zespołu Turnera. Pediatr Endocrinol 2006;5(2):23-30
- 13. Savendahl L, Davenport ML. Delayed diagnosis of Turner’s syndrome: proposed guidelines for change. J Pediatr 2000;137:455-9
- 14. Stephure DK; Canadian Growth Hormone Advisory Committee. Impact of growth hormone supplementation on adult height in turner syndrome: results of the Canadian randomized controlled trial. J Clin Endocrinol Metab 2005;90(6):3360-6
- 15. Ross JL, Quigley CA, Cao D, et al. Growth hormone plus childhood low-dose estrogen in Turner’s syndrome. N Engl J Med 2011;364(13):1230-42
- 16. Darendeliler F, Karagiannis G, Wilton P. Headache, idiopathic intracranial hypertension and slipped capital femoral epiphysis during growth hormone treatment: a safety update from the KIGS database. Horm Res 2007;68(Suppl 5):41-7
- 17. Bell J, Parker KL, Swinford RD, et al. Long-term safety of recombinant human growth hormone in children. J Clin Endocrinol Metab 2010;95(1):167-77
- 18. Bolar K, Hoffman AR, Maneatis T, et al. Long-term safety of recombinant human growth hormone in turner syndrome. J Clin Endocrinol Metab 2008;93(2):344-51
- 19. Tuffli GA, Johanson A, Rundle AC, et al. Lack of increased risk for extracranial, nonleukemic neoplasms in recipients of recombinant deoxyribonucleic acid growth hormone. J Clin Endocrinol Metab 1995;80(4):1416-22
- 20. Menke LA, Sas TC, de Muinck Keizer-Schrama SM, et al. Efficacy and safety of oxandrolone in growth hormone-treated girls with Turner syndrome. J Clin Endocrinol Metab 2010;95(3):1151-60
- 21. Gault EJ, Perry RJ, Cole TJ, et al. Effect of oxandrolone and timing of pubertal induction on final height in Turner’s syndrome: randomised, double blind, placebo controlled trial. BMJ 2011;342:d1980
Następny artykuł:

 Dodaj do ulubionych
Dodaj do ulubionych
