



Spis treści
Streszczenie
Postęp w badaniach nad kontrolą hemostazy ustroju oraz istotne kliniczne implikacje terapeutyczne w odniesieniu do układu krzepnięcia krwi i fibrynolizy wymagają okresowego uaktualniania wytycznych postępowania lekarskiego u chorych z masywną utratą krwi. W niniejszym artykule skupiono się na aspektach klinicznych końcowego etapu formowania skrzepu i aktywności fibrynolitycznej osocza u chorych z masywną utratą krwi. Omówiono mechanizm wczesnej aktywacji białka C, powstawanie
nadmiernej fibrynolizy pokrwotocznej i pourazowej oraz opisano terapię preparatami fibrynogenu u chorych z masywną utratą krwi. Odniesiono się do najnowszych informacji sugerujących farmakologiczne zahamowanie fibrynolizy w okresie okołooperacyjnym i pourazowym. Przedstawione informacje mogą stanowić asumpt do uaktualnienia zaleceń postępowania terapeutycznego z chorymi, u których nastąpiła masywna utrata krwi.
Wprowadzenie
![Tabela. Definicje masywnej utraty krwi wg Stainsby’ego [2] uwzględniające utraconą objętość krwi krążącej i dynamikę utraty](/assets/kv_placeholder_medium-94ce2bba3c62b7bee59dd08888fa63c9f2c3db72880e678190366966855036ea.webp)
Tabela. Definicje masywnej utraty krwi wg Stainsby’ego [2] uwzględniające utraconą objętość krwi krążącej i dynamikę utraty
Zaburzenia krzepnięcia krwi u chorych krwawiących są istotną klinicznie odpowiedzią układu hemostazy na wstrząs. Rozwijające się zaburzenia hemostazy uznawane są za najczęstszą przyczynę wykrwawienia we wczesnym okresie po urazie 4 . Udowodniono, że objętość przetaczanej krwi w okresie pourazowym lub pooperacyjnym jest jednym z najważniejszych czynników rokowniczych przeżycia i częstości wystąpienia powikłań 5 .
Obecnie przyjmuje się, że zapobieganie rozwojowi zaburzeń krzepnięcia krwi może być kluczowym celem terapeutycznym u chorych masywnie krwawiących i przyczynić się do poprawy wyników leczenia. Z chirurgicznego punktu widzenia za najważniejszy element leczenia należy uznać dążenie do powstrzymania krwotoku. Ustalenie takiego priorytetu wymaga przyjęcia szczególnej taktyki postępowania polegającej na stosowaniu uproszczonych technik operacyjnych ograniczających utratę krwi (hemostatic surgery) z następową resuscytacją, wyrównaniem hipotermii i kwasicy oraz wtórnym zabiegami operacyjnymi (stage surgery) 6 . Postępowanie zgodne z tymi zasadami znacznie poprawiło wyniki leczenia chorych z ciężkimi obrażeniami ciała 7, 8, 9, 10 .
Badania nad mechanizmami powstawania zaburzeń krzepnięcia krwi u chorych masywnie krwawiących umożliwiły częściowe poznanie możliwości zapobiegania koagulopatii pourazowej i pokrwotocznej. Wysoki stopień skomplikowania występujących zaburzeń stwarza konieczność prowadzenia terapii wielokierunkowej i multidyscyplinarnej. Dodatkowym zjawiskiem jest fizjologiczna odpowiedź ustroju na niewystarczającą perfuzję tkanek, polegająca na uruchomieniu mechanizmów zapobiegających krzepnięciu przez hamowanie kaskadowej aktywacji osoczowych czynników krzepnięcia i aktywacji fibrynolizy 11 .
Konieczność uporządkowania informacji przedstawianych w piśmiennictwie dała asumpt do stworzenia ogólnych zaleceń postępowania u chorych, u których doszło do masywnej utraty krwi z różnych powodów. W ostatnich latach opisano podstawowe cele terapeutyczne w formie rekomendacji ekspertów europejskich 6 , aktualizowane w miarę postępu prac nad mechanizmami rozwoju zaburzeń krzepnięcia krwi u ofiar ciężkich wypadków 12 . W warunkach polskich przeprowadzono badanie, którego celem było uzyskanie uśrednionej opinii polskich ekspertów dotyczącej zapobiegania i rozpoznawania koagulopatii pokrwotocznej oraz metod wspomagających leczenie masywnej utraty krwi. Wyniki tego badania przedstawiono w formie polskich zaleceń pod auspicjami Stowarzyszenia na Rzecz Leczenia Ciężkich Krwotoków 13 . Obecnie należy przyjąć, że przedstawiane zalecenia europejskie i krajowe częściowo utraciły swą aktualność z powodu ciągłego dostarczania nowych informacji o możliwościach rozpoznawania i leczenia nabytych zaburzeń krzepnięcia krwi.
Mechanizm aktywacji białka C u chorych z niewystarczającą perfuzją tkanek
Poszukiwanie przyczyn zaburzeń krzepnięcia krwi u chorych we wstrząsie umożliwiło postawienie hipotezy o kluczowej roli aktywacji lub uszkodzenia śródbłonka naczyniowego. Dotychczas nie przedstawiono dokładnego mechanizmu aktywacji śródbłonka, przypuszcza się jednak, że zasadniczą rolę odgrywa zmiana architektoniki glikokaliksu spowodowana zmniejszeniem dynamiki przepływu krwi w mikrokrążeniu 14 . Obecnie nie można wykluczyć autoheparynizacji w następstwie uszkodzenia glikokaliksu i uwolnienia wielocukrów o działaniu zbliżonym do heparyny 15 . Skutkiem aktywacji śródbłonka naczyń jest również nadekspresja trombomoduliny wiążącej powstającą w miejscu uszkodzenia naczyń trombinę, następnie aktywacja białka C i zahamowanie kaskady krzepnięcia krwi. U chorych z rozwiniętymi zaburzeniami krzepnięcia krwi w okresie pourazowym stwierdzono zwiększone stężenie trombomoduliny. Stężenie trombomoduliny korelowało też ze śmiertelnością po urazach 11 . Zwiększeniu stężenia trombomoduliny towarzyszyło zmniejszenie stężenia nieaktywnej formy białka C. Niezależnie od innych czynników, zjawisko to wiązało się ze zwiększeniem śmiertelności chorych bezpośrednio po urazie 11 .
Rycina 1. Podstawowe mechanizmy fibrynolizy z uwzględnieniem niektórych aktywatorów i inhibitorów aktywacji plazminy. PAI-1 i PAI-2 – inhibitor aktywatora plazminogenu typu 1 i 2, t-PA i u-PA – tkankowe aktywatory plazminy, odpowiednio tkankowy i urokinaza, HMWK – wysokocząsteczkowy kininogen, FDP – produkty degradacji fibrynogenu.
Nadmierna fibrynoliza
Sprawność układu fibrynolitycznego umożliwia fizjologiczną kontrolę ustroju nad rozmiarem tworzących się skrzeplin oraz odpowiada za przywrócenie przepływu w uszkodzonym naczyniu (ryc. 1). Wielokrotnie udowodniono znaczenie nadmiernej fibrynolizy w rozwoju pourazowych zaburzeń krzepnięcia krwi 16 . Trwają liczne badania mające na celu poznanie szczegółowych mechanizmów odpowiedzialnych za pobudzenie układu fibrynolitycznego 17 . Interesujące są dowody na udział plazminy i jej aktywatorów w regulacji aktywności metaloproteinaz tkankowych (matrix metalloproteinase, MMP), ich inhibitorów (tissue inhibitor of metalloproteinase, TIMP) oraz receptorów lipoprotein o małej gęstości (lipoprotein receptor, LPR) odpowiadających za funkcję bariery krew-mózg 18, 19 . Stwierdzono, że nadmierna fibrynoliza jest ważnym następstwem wstrząsu 20 , koagulopatii położniczej 21 , ciężkich urazów 22 , niewydolności wątroby 23 , fazy anhepatycznej przeszczepienia wątroby 24 , uszkodzenia ośrodkowego układu nerwowego 25 oraz stosowania krążenia pozaustrojowego 26 . Przypuszcza się, że farmakologiczne ograniczenie fibrynolizy może zmienić częstość występowania jawnych koagulopatii 16 , zmniejszyć konieczność przetaczania preparatów krwi oraz poprawić dotychczasowe wyniki leczenia 27 . W badaniu przeprowadzonym z udziałem chorych, którzy doznali ciężkich obrażeń ciała, zastosowanie kwasu traneksamowego pozwoliło na zmniejszenie całkowitej śmiertelności pourazowej o 9% oraz zmniejszenie śmiertelności zależnej od wykrwawienia o 15% w porównaniu z grupą kontrolną otrzymującą placebo 28 . Wykazano też zmniejszenie śmiertelności po leczeniu kwasem traneksamowym chorych z krwawieniem do przewodu pokarmowego. Wyniki metaanalizy 29 wskazują, że stosowanie kwasu traneksamowego zmniejsza śmiertelność o 39% w porównaniu z obserwowaną wśród chorych otrzymujących placebo. W podgrupach chorych leczonych technikami endoskopowymi lub antagonistami receptorów histaminowych typu 2 (cymetydyna) albo inhibitorami pompy protonowej (lanzoprazol) nie stwierdzono korzyści terapeutycznych w porównaniu z obserwowanymi w grupie chorych otrzymujących wyłącznie lek antyfibrynolityczny 29 . Zastosowanie kwasu traneksamowego ogranicza częstość przetaczania krwi u kobiet z masywnymi krwawieniami miesiączkowymi i wykazuje przewagę nad niesteroidowymi lekami przeciwzapalnymi, progestagenami oraz etamsylatem 30 . Nie stwierdzono natomiast wpływu kwasu traneksamowego na zmniejszenie częstości przetaczania krwi u chorych poddawanych przeszczepieniu wątroby 31 . W metaanalizie danych pochodzących od 1913 chorych uczestniczących 33 badaniach wskazano jednak, że rozpoznanie hiperfibrynolizy, jej osłabienie za pomocą aprotyniny lub ograniczenie jej następstw dzięki podaniu rekombinowanego aktywnego czynnika krzepnięcia VII (rFVIIa) prawdopodobnie zmniejsza nasilenie krwawienia i ogranicza zużycie preparatów krwi 31 .
Stężenie fibrynogenu
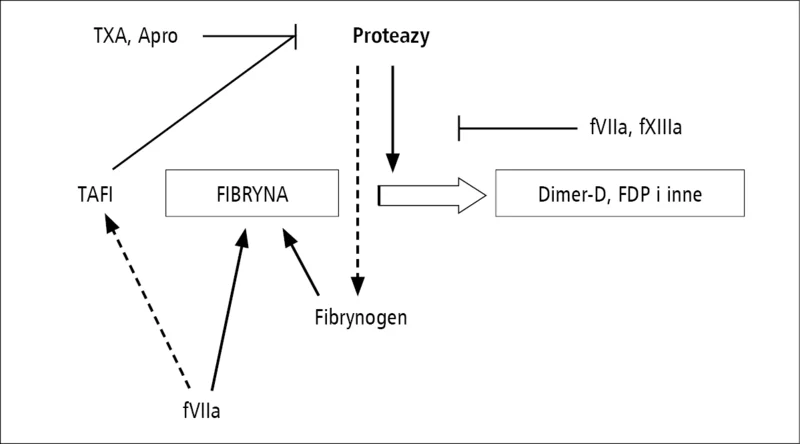
Rycina 2. Wybrane drogi stabilizacji fibryny. TXA – kwas traneksamowy, Apro – aprotynina, fVIIa – aktywny VII czynnik krzepnięcia, fXIIIa – aktywny XIII czynnik krzepnięcia, TAFI – inhibitor fibrynolizy aktywowany przez trombinę, FDP – produkty degradacji fibrynogenu.
Najczęściej wykorzystywane źródła egzogennego fibrynogenu to świeżo mrożone osocze (fresh frozen plasma, FFP), krioprecypitat (KRIO) i liofilizowane koncentraty fibrynogenu ludzkiego (FIB) 39 . Zawartość fibrynogenu w tych preparatach jest zróżnicowana, co utrudnia prowadzenie precyzyjnej terapii substytucyjnej. Ich przetaczanie stwarza również ryzyko przenoszenia wirusów, a w przypadku FFP wystąpienia ostrego potransfuzyjnego uszkodzenia płuc i hiperwolemii 40 . W przeciwieństwie do FFP i KRIO FIB zawiera wystandaryzowaną ilość fibrynogenu, jest bezpieczniejszy pod względem wirusologicznym i gotowy do użycia w ciągu kilku minut. W ostatnich latach wzrasta wykorzystywanie FIB w nabytych hipofibrynogenemiach pourazowych i pooperacyjnych 41 , co pozwala zmniejszyć objętość przetaczanych płynów i ryzyko przenoszenia zakażeń wirusowych.
Udział czynnika XIII stabilizującego fibrynę
Ostateczna stabilizacja skrzepu krwi polega na wytworzeniu wiązań krzyżowych między grupami karboksylowymi glutaminy a resztami aminowymi lizyny sąsiadujących włókien fibryny i przebiega w obecności aktywnego czynnika XIII (XIIIa) oraz jonów wapnia 42 . Rola czynnika XIII nie została do końca poznana, a jego działanie jest bardziej złożone 43 . Pod wpływem czynnika XIIIa stabilny skrzep wiąże do włókien fibryny białkowe inhibitory plazminy (zwłaszcza alfa 2-antyplazminę), zmniejszając podatność skrzepu na działanie proteolityczne 44 . Retrakcja jako końcowy etap dojrzewania skrzepu przebiega w mechanizmie wytwarzania wiązań krzyżowych między białkami szkieletu komórkowego krwinek płytkowych, czyli aktyny, miozyny, filaminy i winkuliny 44 . Udział czynnika XIIIa w wytwarzaniu wewnątrzkomórkowych wiązań krzyżowych między fragmentami endoplazmatycznymi receptorów angiotensyny typu 1 znajdujących się na monocytach jest rozważany jako prawdopodobny mechanizm rozwoju miażdżycy 45 . Obecność czynnika XIII w przestrzeni pozanaczyniowej i macierzy międzykomórkowej umożliwia tworzenie wiązań krzyżowych białek macierzy (fibronektyna, kolagen i czynnik von Willebranda), uczestnicząc tym samym w procesie gojenia ran 43 .
Istnieją podstawy patofizjologiczne uzasadniające słuszność stwierdzenia, że zastosowanie czynnika XIII może ograniczyć procesy fibrynolizy i zwiększyć szansę na uzyskanie stabilnego skrzepu. Pewną przesłanką może być kliniczna skuteczność czynnika XIII w leczeniu krwawień u chorych na hemofilię A 46 oraz skuteczność terapeutyczna czynnika VIIa u krwawiącej osoby z wrodzonym niedoborem czynnika XIII i chorobą Henocha-Schoenleina 47 .
Podsumowanie
Na podstawie przedstawionych danych należy stwierdzić, że ustalanie celów terapeutycznych u chorych z masywną utratą krwi, zogniskowanych na zapobieganiu i leczeniu rozwijających się zaburzeń krzepnięcia, wymaga uzyskania precyzyjnych danych klinicznych i laboratoryjnych. Obecnie uważa się, że przesiewowe testy koagulometryczne nie wystarczają do podjęcia ostatecznych decyzji klinicznych ze względu na trudności interpretacyjne złożonych zaburzeń hemostazy 8 . Dlatego pojawia się skłonność do sięgania po unowocześnione globalne testy układu hemostazy oparte o metody tromboelastometryczne 48, 49 . Na tej podstawie stwierdzono, że u chorych z masywną utratą krwi można obserwować zdolność do nadkrzepliwości, trudności w formowaniu skrzepu oraz cechy nadmiernej fibrynolizy 50 . Ujawniające się zaburzenia zależą od ciężkości doznanego urazu, rozległości uszkodzeń tkankowych oraz zaburzeń perfuzji tkankowej w trakcie krwotoku i okresie pokrwotocznym 16, 50, 51, 52 . Można przyjąć, że rodzaj występujących zaburzeń ma decydujące znaczenie w wyborze celowanej terapii minimalizującej objawy skazy krwotocznej oraz umożliwia zapobieganie wtórnym zakrzepicom.
- 1. Mannucci PM, Levi M. Prevention and treatment of major blood loss. N Engl J Med 2007;356(22):2301-2311.
- 2. Stainsby D, MacLennan S, Hamilton PJ. Management of massive blood loss: a template guideline. Br J Anaesth 2000;85(3):487-491.
- 3. Gutierrez G, Reines HD, Wulf-Gutierrez ME. Clinical review: hemorrhagic shock. Crit Care 2004;8(5):373-381.
- 4. Hoyt DB, Bulger EM, Knudson MM, et al. Death in the operating room: an analysis of a multi-center experience. J Trauma 1994;37(3):426-432.
- 5. Como JJ, Dutton RP, Scalea TM, et al. Blood transfusion rates in the care of acute trauma. Transfusion 2004;44(6):809-813.
- 6. Spahn DR, Cerny V, Coats TJ, et al. Management of bleeding following major trauma: a European guideline. Crit Care 2007;11(1):R17.
- 7. Johnson JW, Gracias VH, Schwab CW, et al. Evolution in damage control for exsanguinating penetrating abdominal injury. J Trauma 2001;51(2):261-269.
- 8. Lynn M, Jeroukhimov I, Klein Y, et al. Updates in the management of severe coagulopathy in trauma patients. Intensive Care Med 2002;28(Suppl 2):S241-S247.
- 9. Moore EE. Thomas G. Orr Memorial Lecture. Staged laparotomy for the hypothermia, acidosis, and coagulopathy syndrome. Am J Surg 1996;172(5):405-410.
- 10. Rotondo MF, Schwab CW, McGonigal MD, et al. 'Damage control': an approach for improved survival in exsanguinating penetrating abdominal injury. J Trauma 1993;35(3):375-382.
- 11. Brohi K, Cohen MJ, Ganter MT, et al. Acute traumatic coagulopathy: initiated by hypoperfusion: modulated through the protein C pathway? Ann Surg 2007;245(5):812-818.
- 12. Rossaint R, Bouillon B, Cerny V, et al. Management of bleeding following major trauma: an updated European guideline. Crit Care 2010;14(2):R52.
- 13. Paluszkiewicz P, Mayzner-Zawadzka E, Baranowski W, et al. Recommendations for the management of trauma or surgery-related massive blood loss. Pol Przegl Chir 2011;83(8):465-476.
- 14. Johansson PI, Stensballe J, Rasmussen LS, et al. A high admission syndecan-1 level, a marker of endothelial glycocalyx degradation, is associated with inflammation, protein C depletion, fibrinolysis, and increased mortality in trauma patients. Ann Surg 2011;254(2):194-200.
- 15. Ostrowski SR, Johansson PI. Endothelial glycocalyx degradation induces endogenous heparinization in patients with severe injury and early traumatic coagulopathy. J Trauma Acute Care Surg 2012;73(1):60-66.
- 16. Ives C, Inaba K, Branco BC, et al. Hyperfibrinolysis elicited via thromboelastography predicts mortality in trauma. J Am Coll Surg 2012;215(4):496-502.
- 17. Lacroix R, Gnat-George F. Microparticles: New Protagonists in Pericellular and Intravascular Proteolysis. Semin Thromb Hemost 2013 (ahead of print).
- 18. Ortolano S, Spuch C. tPA in the Central Nervous System: Relations Between tPA and Cell Surface LRPs. Recent Pat Endocr Metab Immune Drug Discov 2012 (ahead of print).
- 19. Sun HY, Szlam F, Levy JH, et al. Antifibrinolytic agents reduce tissue plasminogen activator-mediated neuronal toxicity in vitro. Acta Anaesthesiol Scand 2009;53(3):325-331.
- 20. Brohi K, Singh J, Heron M, et al. Acute traumatic coagulopathy. J Trauma 2003;54(6):1127-1130.
- 21. Annecke T, Geisenberger T, Kurzl R, et al. Algorithm-based coagulation management of catastrophic amniotic fluid embolism. Blood Coagul Fibrinolysis 2010;21(1):95-100.
- 22. Hess JR, Brohi K, Dutton RP, et al. The coagulopathy of trauma: a review of mechanisms. J Trauma 2008;65(4):748-754.
- 23. Ferro D, Celestini A, Violi F. Hyperfibrinolysis in liver disease. Clin Liver Dis 2009;13(1):21-31.
- 24. Trzebicki J, Kosieradzki M, Flakiewicz E, et al. Detrimental effect of aprotinin ban on amount of blood loss during liver transplantation: single-center experience. Transplant Proc 2011;43(5):1725-1727.
- 25. Goh KY, Tsoi WC, Feng CS, et al. Haemostatic changes during surgery for primary brain tumours. J Neurol Neurosurg Psychiatry 1997;63(3):334-338.
- 26. Kuepper F, Dangas G, Mueller-Chorus A, et al. Fibrinolytic activity and bleeding after cardiac surgery with cardiopulmonary bypass and low-dose aprotinin therapy. Blood Coagul Fibrinolysis 2003;14(2):147-153.
- 27. Shakur H, Roberts I, Piot P, et al. A promise to save 100,000 trauma patients. Lancet 2013;380(9859):2062-2063.
- 28. Shakur H, Roberts I, Bautista R, et al. Effects of tranexamic acid on death, vascular occlusive events, and blood transfusion in trauma patients with significant haemorrhage (CRASH-2): a randomised, placebo-controlled trial. Lancet 2010;376(9734):23-32.
- 29. Gluud LL, Klingenberg SL, Langholz E. Tranexamic acid for upper gastrointestinal bleeding. Cochrane Database Syst Rev 2012;(1):CD006640.
- 30. Lethaby A, Farquhar C, Cooke I. Antifibrinolytics for heavy menstrual bleeding. Cochrane Database Syst Rev 2000;(4):CD000249.
- 31. Gurusamy KS, Pissanou T, Pikhart H, et al. Methods to decrease blood loss and transfusion requirements for liver transplantation. Cochrane Database Syst Rev 2011;(12):CD009052.
- 32. Bolliger D, Gorlinger K, Tanaka KA. Pathophysiology and treatment of coagulopathy in massive hemorrhage and hemodilution. Anesthesiology 2010;113(5):1205-1219.
- 33. Franchini M, Lippi G. Fibrinogen replacement therapy: a critical review of the literature. Blood Transfus 2012;10(1):23-27.
- 34. Schlimp CJ, Cadamuro J, Solomon C, et al. The effect of fibrinogen concentrate and factor XIII on thromboelastometry in 33% diluted blood with albumin, gelatine, hydroxyethyl starch or saline in vitro. Blood Transfus 2012;10(1):1-9.
- 35. Fenger-Eriksen C, Tonnesen E, Ingerslev J, et al. Mechanisms of hydroxyethyl starch-induced dilutional coagulopathy. J Thromb Haemost 2009;7(7):1099-1105.
- 36. Inaba K, Karamanos E, Lustenberger T, et al. Impact of Fibrinogen Levels on Outcomes after Acute Injury in Patients Requiring a Massive Transfusion. J Am Coll Surg 2012 (ahead of print).
- 37. Bolliger D, Szlam F, Molinaro RJ, et al. Finding the optimal concentration range for fibrinogen replacement after severe haemodilution: an in vitro model. Br J Anaesth 2009;102(6):793-799.
- 38. Dempfle CE, Kalsch T, Elmas E, et al. Impact of fibrinogen concentration in severely ill patients on mechanical properties of whole blood clots. Blood Coagul Fibrinolysis 2008;19(8):765-770.
- 39. Fenger-Eriksen C, Ingerslev J, Sorensen B. Fibrinogen concentrate – a potential universal hemostatic agent. Expert Opin Biol Ther 2009;9(10):1325-1333.
- 40. Norda R, Tynell E, Akerblom O. Cumulative risks of early fresh frozen plasma, cryoprecipitate and platelet transfusion in Europe. J Trauma 2006;60(6 Suppl):S41-S45.
- 41. Fries D, Martini WZ. Role of fibrinogen in trauma-induced coagulopathy. Br J Anaesth 2010;105(2):116-121.
- 42. Richardson VR, Schroeder V, Grant PJ, et al. Complement C3 is a substrate for activated factor XIII that is cross-linked to fibrin during clot formation. Br J Haematol 2013;160(1):116-119.
- 43. Richardson VR, Cordell P, Standeven KF, et al. Substrates of Factor XIII-A: roles in thrombosis and wound healing. Clin Sci (Lond) 2013;124(3):123-137.
- 44. Kasahara K, Souri M, Kaneda M, et al. Impaired clot retraction in factor XIII A subunit-deficient mice. Blood 2010;115(6):1277-1279.
- 45. Thomas WG. Double trouble for type 1 angiotensin receptors in atherosclerosis. N Engl J Med 2005;352(5):506-508.
- 46. Rea CJ, Foley JH, Sorensen B. Factor XIII in the treatment of hemophilia A. N Engl J Med 2012;366(3):281-283.
- 47. Alioglu B, Ozsoy MH, Tapci E, et al. Successful use of recombinant factor VIIa in a child with Schoenlein-Henoch purpura presenting with compartment syndrome and severe factor XIII deficiency. Blood Coagul Fibrinolysis 2013;24(1):102-105.
- 48. Schreiber MA. The beginning of the end for damage control surgery. Br J Surg 2012;99 Suppl:110-11.
- 49. Sorensen B, Fries D. Emerging treatment strategies for trauma-induced coagulopathy. Br J Surg 2012;99(Suppl):140-50.
- 50. Ostrowski SR, Sorensen AM, Larsen CF, et al. Thrombelastography and biomarker profiles in acute coagulopathy of trauma: a prospective study. Scand J Trauma Resusc Emerg Med 2011;19:64.
- 51. Fenger-Eriksen C, Jensen TM, Kristensen BS, et al. Fibrinogen substitution improves whole blood clot firmness after dilution with hydroxyethyl starch in bleeding patients undergoing radical cystectomy: a randomized, placebo-controlled clinical trial. J Thromb Haemost 2009;7(5):795-802.
- 52. Martini WZ, Cortez DS, Dubick MA, et al. Thrombelastography is better than PT, aPTT, and activated clotting time in detecting clinically relevant clotting abnormalities after hypothermia, hemorrhagic shock and resuscitation in pigs. J Trauma 2008;65(3):535-543.
Pierwszy artykuł:

 Dodaj do ulubionych
Dodaj do ulubionych
