


Co znajdziesz w artykule?
- Trichoblastoma – łagodny nowotwór pochodzenia włosowego
- Obraz kliniczny postaci pojedynczych i mnogich
- Aktualne metody diagnostyki i leczenia
Spis treści
Trichoblastoma (TB) to łagodny nowotwór pochodzenia włosowego o dwoistym zróżnicowaniu: z rozrodczego nabłonka mieszkowego i specyficznego mieszkowego zrębu. Charakterystycznym elementem histologicznym są tzw. papillary mesenchymal bodies – struktury przypominające brodawkę włosa anagenowego. Najczęściej TB występują jako pojedyncze guzy sporadyczne. Mogą także rozwijać się jako guzy wtórne na podłożu znamienia łojowego Jadassohna. Mnogie TB są natomiast najczęściej związane z zespołem
Brooke’a-Spieglera, obecnie zaliczanym do skórnych zespołów zależnych od mutacji w genie CYLD, rzadziej stanowią element innych zespołów 1 .
Termin „trichoblastoma” został wprowadzony w 1970 r. przez Headingtona, a następnie zaadaptowany do określenia całej grupy nowotworów, obejmującej także guzy określane wcześniej mianem „trichoepithelioma” (TE; polska nazwa: nabłoniak włosowy). Uznano, że nazwa TB lepiej oddaje ich histogenezę. Obecnie nazwy TE używa się zatem tylko dla określenia niektórych typów histologicznych TB: cribriform TB (ogniska nowotworowe przypominające sito) ➞ klasyczny TE; columnar TB (ogniska nowotworowe układające się w wąskie kolumny) ➞ desmoplastyczny TE; występuje ona także w określeniu zespołu „multiple familial trichoepithelioma” (MFT; polska nazwa: zespół mnogich rodzinnych nabłoniaków włosowych), w którym stwierdza się mnogie TB, najczęściej typu klasycznego TE 2 .
Odmiany kliniczne
Sporadyczne, pojedyncze TB
Pojawiają się najczęściej w 5-6 dekadzie życia w okolicy głowy i szyi. Są to zazwyczaj asymptomatyczne, kopulaste, spoiste, cieliste lub różowe guzki bądź guzy, czasem o teleangiektatycznej powierzchni, o średnicy 0,5-3 cm. Guzy mogą zawierać pigment (dotyczy to zwłaszcza dużych zmian) 2 .
Plaque-type TB
Jest to rzadka odmiana tego rodzaju guza, występująca jako pojedyncza lub mnoga, słabo odgraniczona guzkowa tarczka, cechująca się miejscową złośliwością, mająca tendencję do naciekania głębszych struktur i z tego względu wymagająca często reoperacji 3 .
Desmoplastyczny TE
Występuje najczęściej na twarzy w postaci asymptomatycznej porcelanowej blaszki o średnicy ok. 1 cm, z lekko uniesionym brzegiem i zagłębionym centrum (ryc. 1). Zmiana najczęściej jest pojedyncza, rzadziej mnoga 2 .

Rycina 1. Desmoplastyczny nabłoniak włosowy (desmoplastic trichoepithelioma, columnar trichoblastoma) u kobiety w średnim wieku
TB w obrębie znamienia łojowego Jadassohna
Znamię łojowe opisane po raz pierwszy w 1895 r. przez Jadassohna to hamartoma lub inaczej znamię organoidne, w którym nieprawidłowości dotyczą naskórka, gruczołów łojowych, innych gruczołów związanych z mieszkiem włosowym oraz skóry, powstające w wyniku mozaicyzmu genetycznego w życiu płodowym. Znamię to występuje najczęściej w obrębie głowy i szyi, zwłaszcza na owłosionej skórze głowy, rzadziej w okolicach genitalnych, od urodzenia w postaci pojedynczej, pozbawionej włosów tarczki lub pasma o woskowatej, żółtawej, lekko wyboistej lub (w przypadku większych znamion) mózgowatej powierzchni. W takiej postaci utrzymuje się do okresu pokwitania, kiedy to staje się bardziej wyniosłe, brodawkowate i przybiera ciemniejszą, brunatnożółtawą barwę, zależną od stymulowanego androgenami rozrostu gruczołów łojowych. W 4 i 5 dekadzie życia w 10-20% przypadków w obrębie znamienia mogą rozwijać się wtórne guzy pochodzenia naskórkowego, przydatkowego lub mezenchymalnego. W większości są to guzy łagodne: TB, syringocystadenoma papilliferum (SCAP), tricholemmoma, proliferating trichilemmal tumor, pilomatricoma, tumor of the follicular infundibulum, sebaceoma. Dużo rzadziej zdarzają się guzy złośliwe: rak podstawnokomórkowy (BCC – basal cell carcinoma), rak łojowy, rak płaskonabłonkowy rogowaciejący, rak z gruczołów apokrynowych czy syringocystadenocarcinoma papilliferum.
Podejrzenie TB budzi pojawiający się w dojrzałym wieku, nowy, odbiegający strukturą od pozostałej części znamienia, spoisty guz o gładkiej powierzchni, często przebarwiony. Owrzodzenie lub rozpad stwierdza się rzadziej niż w innych wtórnych guzach, co różnicuje go klinicznie od SCAP oraz nowotworów złośliwych. Przybiera on najczęściej postać dużego guza (large nodular variant), ale inne warianty histologiczne TB (drobnoguzkowy, cribriform, retiform czy racemiform) także były opisywane 2 .
Mnogie TB jako element zespołów genetycznych
Zespół Brooke’a-Spieglera (BSS – Brooke-Spiegler syndrome – OMIM 605041) jest chorobą dziedziczoną autosomalnie dominująco, charakteryzującą się tendencją do występowania mnogich łagodnych skórnych guzów przydatkowych: cylindroma, spiradenoma, spiradenocylindroma i TE (cribriform TB) oraz rzadziej guzów gruczołów ślinowych, najczęściej membranous basal cell adenoma (MBCA), a także guzów w płucach – pulmonary cylindromas 4 . Wymienione guzy zwykle występują w określonych lokalizacjach, a mianowicie: cylindroma na głowie i szyi, TE w środkowej części twarzy, spiradenoma na tułowiu. Guzy mogą pojawiać się synchronicznie lub metachronicznie, może także występować więcej niż jeden typ nowotworów wymienionych powyżej. Według niektórych autorów współistnienie cylindroma/spiradenoma/trichoepithelioma stwierdzano u ok. 15% pacjentów 5 .
U części chorych występuje wyłącznie jeden typ mnogich guzów: cylindroma (oblak) lub TE, a schorzenia te określa się odpowiednio rodzinną cylindromatozą (FC – familial cylindromatosis – OMIM 132700) oraz zespołem mnogich rodzinnie występujących nabłoniaków włosowych (MFT1 – multiple familial trichoepitheliomas-1 – OMIM 601606). W tych jednostkach chorobowych mimo różnic fenotypowych udowodniono obecność mutacji genetycznych w obrębie genu CYLD zlokalizowanego na chromosomie 16q12-q13. Grupę chorób dermatologicznych uwarunkowanych mutacjami w obrębie tego genu, powodującymi utratę funkcji, nazwano zespołem skórnym CYLD (CCS – CYLD cutaneous syndrome). Jednym z dowodów, że wymienione jednostki chorobowe stanowią fenotypowe warianty tego samego zespołu, jest występowanie BSS, FC lub MFT1 u członków tej samej rodziny 4, 5 . Za odmianę BSS uważa się również opisany w 1975 r. zespół Rasmussena jako kombinację mnogich TE, cylindroma i prosaków 2 .
U części pacjentów prezentujących fenotypowe objawy zespołu mnogich rodzinnie występujących nabłoniaków włosowych udowodniono obecność mutacji genetycznych w chromosomie 9p21 (gen PTCH1). Te przypadki określa się jako MFT2 (szczegółowe dane dotyczące występowania, diagnostyki, leczenia itd. tego schorzenia obecnie nie są dostępne), w odróżnieniu od wcześniej wspomnianych MFT1 z mutacją w genie CYLD 2, 6, 7 . Poza tym opisano kilka przypadków mnogich TE niewystępujących rodzinnie (MNF-TE – multiple non-familial TE), przy czym w jednym z nich zmiany były zlokalizowane tylko po prawej stronie twarzy, co powinno nasuwać podejrzenie mozaicyzmu 4, 8, 9 .
Objawy skórne w CCS zwykle pojawiają się w 2 lub 3 dekadzie życia. Pierwsze zmiany zazwyczaj rozwijają się w okresie dojrzewania na skórze głowy i twarzy, ale mogą również występować na tułowiu i w miejscach chronionych przed słońcem, takich jak skóra narządów płciowych i pach. W kolejnych latach zmiany stają się liczniejsze, większe i mogą tworzyć skupiska. Nie mają tendencji do ustępowania. U kobiet zmiany często są bardziej liczne niż u mężczyzn 5 .
Pierwsze TE (cribriform TB) pojawiają się już w dzieciństwie, a w okresie pokwitania i w kolejnych okresach życia guzków przybywa (z czasem ich liczba sięga od kilku do kilkudziesięciu, a nawet kilkuset). Lokalizują się najczęściej obustronnie w okolicach fałdów nosowo-policzkowych (ryc. 2), przyśrodkowych części powiek oraz na pozostałej skórze twarzy, a nawet w przewodach słuchowych zewnętrznych (ryc. 3) w postaci drobnych (0,2-1 cm), kopulastych, spoistych, cielistych guzków, czasem tworzących zlewne konglomeraty. Nie mają tendencji do wrzodzenia, a jeśli taka wystąpi, to (pomijając uraz mechaniczny) może wskazywać na pojawienie się zmiany złośliwej, zwłaszcza BCC 2 .
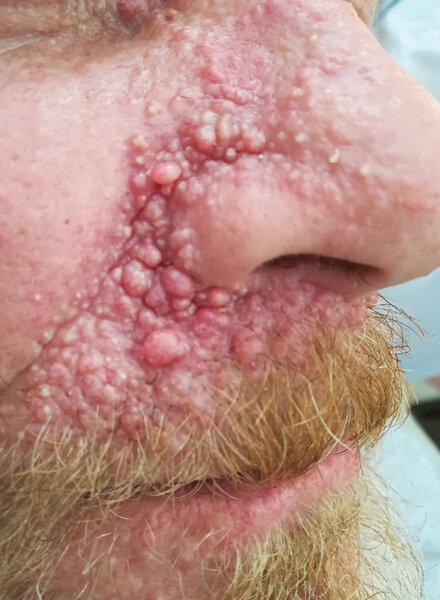
Rycina 2. Liczne nabłoniaki włosowe (klasyczne trichoepithelioma, cribriform trichoblastoma) w okolicach fałdu nosowo-policzkowego, górnej wargi oraz przyśrodkowych części powiek w zespole mnogich rodzinnych nabłoniaków włosowych (multiple familial trichoepithelioma) u mężczyzny w średnim wieku
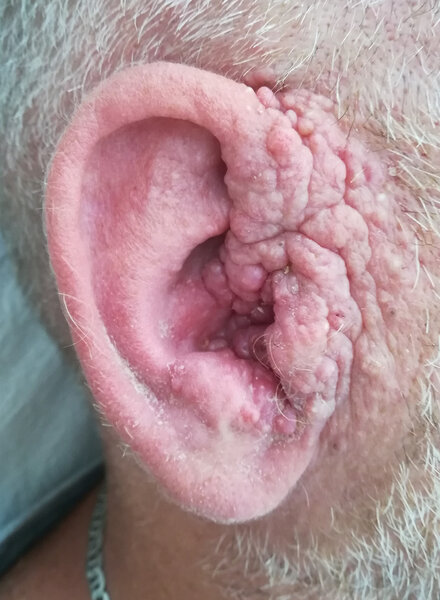
Rycina 3. Konglomerat nabłoniaków włosowych w obrębie małżowiny usznej powodujący osłabienie słuchu u tego samego mężczyzny
Oblak (cylindroma) jest najczęstszym nowotworem w BSS. Występuje on w obrębie skalpu i okolic uszu w postaci wolno rosnących, spoistych guzów o gładkiej, różowej lub niebieskawej, czasem teleangiektatycznej powierzchni, o średnicy 0,5-3 cm, zazwyczaj licznych (od dziesięciu do kilkuset), tworzących konglomeraty – co było powodem określenia mnogich cylindroma mianem „guza turbanowego” (turban tumor). Również spiradenoma występuje w postaci różowych lub niebieskawych guzków o gładkiej powierzchni, głównie na tułowiu, ale też poza nim, niekiedy jest bolesny. Oba guzy są klinicznie podobne, różnią się jednak obrazem histologicznym (choć czasem prezentują cechy wspólne, co jest określane mianem „spiradenocylindroma” i stanowi kolejny dowód przemawiający za ich wspólnym rodowodem). Histologicznie mają one cechy zbliżone do gruczołów potowych apokrynowych, jednak histogenetycznie należy je wiązać z szerzej pojętą jednostką włosowo-łojowo-apokrynową. Mimo że te nowotwory zwykle są łagodne, mogą ulec transformacji złośliwej, którą należy podejrzewać w przypadku zmian różniących się wyglądem od innych, rosnących w szybkim tempie, a także pojawienia się krwawienia lub owrzodzenia, co występuje w 5-10% przypadków 2, 5 .
U niektórych osób z CCS (ok. 3%) rozwijają się nowotwory gruczołów ślinowych (MBCA), zwykle po 40 r.ż., w śliniance przyusznej; mogą być obustronne. MBCA jest chorobą łagodną, którą można leczyć chirurgicznie, ale u 25% chorych guzy nawracają. Jeszcze rzadziej (dotychczas opisano 3 przypadki) występuje cylindroma płuc. Guzy te, pojedyncze lub mnogie, mogą rozwijać się w dużych drogach oddechowych i utrudniać oddychanie 4 .
CCS jest rzadkim zespołem, choć dokładna częstość występowania nie jest znana. Jego występowanie stwierdza się we wszystkich grupach etnicznych, dotyka on w równym stopniu kobiet, jak i mężczyzn. Badania genetyczne w kierunku mutacji CYLD powinno się rozważyć u pacjentów, u których ≥2 cylindroma, spiradenoma lub TE zostały potwierdzone histopatologicznie, oraz w przypadku stwierdzenia pojedynczych cylindroma, spiradenoma lub TE u pacjentów mających krewnych pierwszego stopnia, u których histologicznie potwierdzono którykolwiek z tych nowotworów 5 .
TB opisywano także jako składowe innych niż CCS zespołów genetycznych, do których zalicza się:
- Zespół Bazex-Dupré-Christol (OMIM 301845) – do najczęściej opisywanych w piśmiennictwie cech fenotypowych tego schorzenia należą zaburzenia struktury włosów (pili torti i/lub trichorrhexis nodosa), prosaki oraz skłonność do występowania BCC w późniejszych okresach życia, ale także zanikowe zapalenie mieszków włosowych, TE, hypotrichosis i hypohidrosis. Mutacja odpowiedzialna za ten zespół jest dziedziczona w sposób dominujący i została zlokalizowana na długim ramieniu chromosomu X 8, 10, 11 .
- Zespół Rombo – składają się na niego zanik mieszków włosowych, atrophoderma vermiculatum, zanik brwi i rzęs, hypotrichosis, hypohidrosis, sinica palców rąk i stóp, prosaki, TE, BCC. Jest dziedziczony autosomalnie dominująco 2 .
Obraz dermoskopowy TB
Główną cechą TB obserwowaną w dermatoskopii są krótkie, rozgałęziające się teleangiektazje, czasem tworzące koronę na obwodzie. Tło jest perłowe, z prześwitującymi prosakami, białawymi pasmami i brązowymi globulami. W klasycznym TE (a więc tym spotykanym w zespołach genetycznych) obraz jest podobny: na tle białego podłoża z prześwitującymi prosakami widać drobne, cienkie, rozgałęzione naczynia. W desmoplastycznym TE podłoże ma kolor kości słoniowej lub perłowobiały, a naczynia mogą być grubsze i bardziej rozgałęzione.
Bardzo podobny dermatoskopowy obraz rozgałęziających się teleangiektatycznych naczyń występuje w przypadku guzkowej postaci BCC oraz guza Pinkusa i choć naczynia są w nich grubsze, bardziej rozgałęzione, to nie na tyle, by w sposób pewny zróżnicować te guzy z TB na podstawie dermatoskopii 12 . Podobnie brązowoszare, owoidne struktury, będące wynikiem gromadzenia melaniny w gniazdach nowotworowych, występują zarówno w TB (zwłaszcza w dużych guzach), jak i w BCC, choć częściej można je znaleźć w przypadku pierwszego z nich, podobnie jak struktury prosakopodobne i żółte globule 13 . Jeszcze trudniejsze wydaje się różnicowanie dermoskopowe TB z BCC w obrębie znamienia łojowego z powodu niewyraźnego wzorca naczyniowego. Pomocne w rozpoznaniu obu guzów mogą tu być czarne lub szarobrązowe owoidne struktury, jednak różnice w obrazie są zbyt dyskretne, aby na nich opierać ostateczną diagnozę 14 . Podstawą różnicowania jest badanie histopatologiczne.
Obraz TB w refleksyjnej mikroskopii konfokalnej
W literaturze są dostępne pojedyncze prace dotyczące przydatności refleksyjnej mikroskopii konfokalnej (RCM – reflectance confocal microscopy) w różnicowaniu TB i BCC. Pampena i wsp. uznali szczeliny okołoguzowe (peritumoural clefting), obecne jedynie w BCC, za cechę kluczową w różnicowaniu tego nowotworu z TB 15 . Jednak brak tych szczelin w badaniu RCM nie przesądza o rozpoznaniu TB 16 .
Histopatologia TB i warianty histologiczne
TB to umiejscowione w skórze właściwej, rzadziej sięgające do tkanki podskórnej, dobrze odgraniczone guzy, czasami tworzące skupisko guzków. Guzy nie mają połączenia z naskórkiem, wyrastają z mieszków włosowych i składają się z dwóch elementów: nabłonkowego, odpowiadającego komórkom zawiązków mieszka włosowego, który tworzą bazaloidne komórki o skąpej cytoplazmie, oraz otaczającego elementu łącznotkankowego, który miejscami odpowiada mezenchymalnemu zrębowi brodawek włosa, z charakterystycznymi puchatymi fibroblastami, w związku z czym można zaobserwować typowe struktury określane jako „papillary mesenchymal bodies” („mezenchymalne ciałka brodawek włosa”), w pozostałych miejscach ogniska nabłonkowe są zaś otoczone delikatnymi włóknami kolagenowymi i cienkimi fibroblastami, co odpowiada włóknistej pochewce włosa (ryc. 4). Mezenchymalne ciałka brodawek włosa mogą przypominać typową brodawkę włosa z wypukłą częścią mezenchymalną otoczoną półkolistym rzędem walcowatych komórek bazaloidnych, ale częściej brodawka jest spłaszczona lub wklęsła, co jest określane jako „continuous follicular papilla” („ciągła brodawka mieszka włosowego”) (ryc. 5). Wśród pasm bazaloidnych komórek można dostrzec drobne torbielki mieszkowe. Mogą one również zawierać komórki jasne zróżnicowane w kierunku komórek tricholemmy lub komórek łojowych. Czasem bywają skolonizowane przez melanocyty i komórki Merkla. Tworzące guz jednostki włóknisto-nabłonkowe są otoczone szczelinami oddzielającymi go od zdrowej skóry. Łącznotkankowy zrąb może w niektórych przypadkach zawierać amyloid, większe ilości mucyny lub komórki tłuszczowe 1, 2 .
Rycina 4. Obraz mikroskopowy nabłoniaka włosowego (klasycznego trichoepithelioma). Ogniska bazaloidnych komórek wyrastające wąskimi pasmami z pochewek mieszków włosowych (typ cribriform i retiform), otoczone włóknistym zrębem ze szczeliną otaczającą guzek (H + E, powiększenie obiektywu 10 ×)
Rycina 5. Obraz mikroskopowy nabłoniaka włosowego (klasycznego trichoepithelioma). „Papillary mesenchymal body” oraz fragment szczeliny w otaczającym guzek zrębie (H + E, powiększenie obiektywu 40 ×)
Ze względu na cechy architektoniczne wyróżnia się następujące odmiany TB:
- large nodular: duży guz lub konglomerat złożony z kilku guzków, często z widocznymi złogami melaniny (częste utkanie w TB na podłożu znamienia łojowego)
- small nodular: drobne guzki zbudowane z elementu nabłonkowego, otoczone włóknistym zrębem, który dominuje w masie nowotworowego guzka
- adamantinoid (lymphadenoma): charakterystyczna jest budowa ognisk nabłonkowych, których obwód tworzy rząd palisadowato ułożonych komórek bazaloidnych, a środek formują większe jasne komórki o pęcherzykowatym jądrze, z wyraźnymi jąderkami i obfitą cytoplazmą, oraz limfocyty, wśród których widać też większe komórki wielojądrzaste podobne do komórek Reed-Sternberga
- retiform, racemiform: ogniska komórek bazaloidnych układem przypominają sieć lub kiście winogron
- cribriform (klasyczny TE): ogniska komórek bazaloidnych przypominają sito
- columnar (desmoplatyczny TE): najczęściej zlokalizowany w górnych warstwach skóry siateczkowatej, tworzą go drobne pasma, kolumny i gniazda bazaloidnych komórek otoczone zbitym włóknistym zrębem (ryc. 6) 2 .
Rycina 6. Obraz mikroskopowy desmoplastycznego nabłoniaka włosowego (H + E, powiększenie obiektywu 10 ×)
Różnicowanie histologiczne obejmuje głównie BCC, który nie ma charakterystycznego dla TB zrębu mieszkowego i brodawkowatych ciałek mezenchymalnych, szczeliny znajdują się tuż pod błoną podstawną ognisk nowotworowych; w większości przypadków wyrasta on z dolnej części naskórka. Drugim podobnym do TB nowotworem, zwłaszcza w przypadku powierzchownych biopsji, jest microcystic adnexal carcinoma (MAC), nowotwór złośliwy o naciekającym wzroście 2 .
Immunohistochemicznie komórki tworzące część nabłonkową guzów mają profil komórek rozrodczych mieszka (follicular germ cells), wykazują ekspresję cytokeratyn CK5/6, CK14, CK15, CK17 i CK19, przy braku ekspresji cytokeratyn 1, 4, 7, 8, 10, 13, 18 i 20. Ponadto są BerEP4 pozytywne. Są to markery typowe również dla BCC, zatem wymienione immunohistochemiczne barwienia nie różnicują tych dwóch podobnych do siebie guzów. Natomiast MAC jest BerEP4 negatywny i ten marker immunohistochemiczny może być przydatny do różnicowania 2 . Markerem immunohistochemicznym różnicującym TB od BCC jest receptor androgenowy (AR) negatywny w TB i pozytywny w BCC 6 .
Składowa mezenchymalna guzów wykazuje ekspresję CD34, smooth muscle actin oraz intermediate filament nestin, nie są one jednak specyficzne dla TB 2 .
Uwzględniając powyższe informacje, przeprowadzanie analizy immunohistochemicznej w celu odróżnienia TB od zmian złośliwych nie jest rutynową praktyką, a morfologia komórek, szczególnie zaś ich konfiguracja, nadal odgrywa fundamentalną rolę w postawieniu właściwej diagnozy 17 .
Ryzyko zezłośliwienia
Ryzyko zezłośliwienia w przypadku guzów sporadycznych jest niskie. Istnieją dwa histologiczne warianty złośliwej formy TB:
- rak trichoblastyczny (TBCA – trichoblastic carcinoma)
- rakomięsak trichoblastyczny (TBCASA – trichoblastic carcinosarcoma).
Pierwszy z nich charakteryzuje się atypią komórkową w obrębie komponentu nabłonkowego i „łagodnym” mieszkowym komponentem zrębowym, w drugim zaś atypia komórkowa dotyczy komórek obu składowych: części nabłonkowej oraz mieszkowego zrębu. Jądra komórek są hiperchromatyczne, nieregularne, występują atypowe figury podziałów oraz tzw. crowding, czyli skupienia atypowych komórek.
W genetycznych zespołach obok TB częściej pojawiają się BCC niż TBCA lub TBCASA, jednak spiradenoma może przejść w spiradenocarcinoma, a oblak w cylindrocarcinoma 2 .
Podłoże genetyczne
W sporadycznych guzach znajdowano mutacje oraz polimorfizmy w genach: CTNNB1 (kodującym β-kateninę), HRAS, PTCH1 (gen supresorowy, którego mutacje odpowiadają m.in. za rozwój BCC).
Mutacje w genie CYLD są odpowiedzialne m.in. za mnogie TE w CCS (MFT1). Obecnie opisano już ponad 100 patologicznych wariantów CYLD i ciągle odkrywa się nowe. Co ważne w kontekście CCS, CYLD negatywnie reguluje kilka kluczowych szlaków wpływających na przeżycie komórek ważnych dla rozwoju, wzrostu i utrzymania włosów, w tym NF-κB, Wnt, Notch i TGF-β, które są również ważne w stanach zapalnych i nowotworach. Zwraca uwagę brak korelacji genotypowo-fenotypowej i wydaje się, że stwierdzenie konkretnej mutacji nie pozwala przewidzieć ciężkości choroby. Oprócz kilku innych przypuszczalnych, ale niezidentyfikowanych przyczyn środowiskowych lub stylu życia, dużą różnorodność fenotypową u pacjentów z CCS można również wyjaśnić obecnością wariantów genetycznych zlokalizowanych poza locus genu CYLD, które mogłyby dodatkowo modyfikować fenotyp. Warianty genetyczne zidentyfikowane w STAT3, TRAF3 i NBR1 przy jednoczesnej utracie funkcji CYLD mogą wpływać na aktywność NF-κB w komórkach nowotworowych w przebiegu CCS – wyniki ostatnich analiz funkcjonalnych potwierdzają rolę tych genów w modulacji funkcji CYLD 4 .
W MFT2 mutacje nie dotyczą genu CYLD, lecz PTCH1 2, 4, 5, 7 .
Diagnostyka różnicowa
W różnicowaniu klinicznym pojedynczych TB należy wziąć pod uwagę BCC, znamię Mieschera oraz inne łagodne guzy przydatkowe: torbiel naskórkową, torbiel skórzastą, torbiel włosową, oblaka (cylindroma), eccrine poroma, eccrine nevus, znamię zaskórnikowe, gruczolaka łojowego, trichofolliculoma, tricholemmoma, nabłoniaka wapniejącego (epithelioma calcificans Malherbe, pilomatrixoma).
Desmoplastyczny TE może przypominać twardzinopodobnego BCC lub zanikową bliznę.
Przy zmianach mnogich TE mogą przypominać wiele innych chorób, w przebiegu których występują liczne zmiany grudkowo-guzowate zlokalizowane przede wszystkim w obrębie twarzy. Zalicza się do nich: prosaki, mięczaka zakaźnego, wykwity trądzikowe, przerost gruczołów łojowych, gruczolaka potowego przewodowego (syringoma), torbielaki potowe (eccrine hidrocystoma). Nie należy zapominać, że niektóre z wymienionych chorób mogą występować równocześnie z TE.
Mnogie TB, zwłaszcza jeśli towarzyszą im guzy w innych lokalizacjach, wymagają różnicowania z takimi zespołami genetycznymi, jak:
- Nerwiakowłókniakowatość typu 1 (choroba von Reckling-hausena) – choroba genetyczna o dziedziczeniu autosomalnie dominującym, z mutacją genu NF1, który jest zlokalizowany na chromosomie 17 (17q11.2). Nerwiakowłókniaki – gładkie, cieliste guzy/guzki – są obok plam café-au-lait najczęściej występującym objawem. Mogą pojawiać się u chorych w każdym wieku, a ich liczba i wielkość wraz z wiekiem rośnie. Występują na twarzy, tułowiu i innych częściach ciała. W odróżnieniu od TE są miękkie.
- Stwardnienie guzowate (TSC – tuberous sclerosis complex, choroba Bourneville’a-Pringle’a) – jest chorobą genetyczną dziedziczoną autosomalnie dominująco. W przypadku tego schorzenia występuje mutacja w genie TSC1 (stwardnienie guzowate 1) lub TSC2 (stwardnienie guzowate 2), która prowadzi do wzrostu hamartoma w wielu narządach (mózgu, sercu, płucach, narządzie wzroku, nerkach, kościach). Zmiany skórne obejmują m.in. liczne naczyniakowłókniaki – guzki występujące na twarzy w lokalizacji podobnej do MFT, jednak drobniejsze i bardziej czerwone (czasami przypominające trądzik). Inne skórne objawy to znamiona łącznotkankowe (zwane skórą szagrynową), odbarwione plamy w kształcie liścia i występujące na palcach guzki Koenena oraz włókniaki lub przerost dziąseł.
- Zespół Birta-Hogg-Dubégo (znany również jako zespół Hornsteina-Knickenberg) – jest rzadką genodermatozą dziedziczoną autosomalnie dominująco. Charakteryzuje się obecnością licznych fibrofolliculoma (cielistych guzków na twarzy, głowie i górnej części tułowia, o średnicy 1-5 mm), torbieli płucnych (z odmą opłucnową lub bez niej) oraz zwiększoną podatnością na nowotwory nerek, które mogą występować obustronnie.
- Zespół Cowden – uwarunkowany patogennymi wariantami linii zarodkowej w genie PTEN, zlokalizowanym na chromosomie 10q22-23. Charakteryzuje się makrocefalią, zmianami śluzówkowo-skórnymi (tricholemmoma twarzy, rogowacenie dłoni i stóp oraz grudki brodawkowate), a także występowaniem łagodnych i złośliwych nowotworów piersi, tarczycy, endometrium i nerek.
- Zespół Muira-Torre’a (MTS – Muir-Torre syndrome) – to kliniczna odmiana zespołu Lyncha, charakteryzująca się synchronicznym lub metachronicznym występowaniem co najmniej jednego z nowotworów łojowych (sebaceous adenoma, sebaceous epithelioma, sebaceous carcinoma) lub keratoacanthoma i co najmniej jednego nowotworu narządów wewnętrznych.
- Zespół Carneya – to rzadka choroba genetyczna związana z zespołem mnogiej gruczolakowatości wewnątrzwydzielniczej. Śluzaki skóry (myxoma) – drobne, płaskie albo większe, uszypułowane, opalizujące lub ciemnoróżowe, asymptomatyczne guzki – są najbardziej swoistym objawem dermatologicznym, pozwalającym na rozpoznanie zespołu Carneya (stwierdza się je u 20-55% pacjentów). Mogą wystąpić w dowolnym miejscu, ale zwykle dotyczą powiek, uszu, sutków, zewnętrznego przewodu słuchowego, tułowia i krocza. Inne odchylenia dermatologiczne to plamy soczewicowate, znamiona błękitne i plamy typu café-au-lait.
- Zespoły Bazex-Dupré-Christol i Rombo opisano we wcześniejszej części tekstu 2 .
Profilaktyka
W przypadku zmian sporadycznych brak zaleceń dotyczących profilaktyki.
Przy zmianach mnogich należy unikać promieniowania UV. W części prac zaleca się także unikanie radioterapii, ponieważ powoduje ona uszkodzenie DNA i może skutkować dalszym powstawaniem nowotworów lub złośliwą transformacją istniejących zmian. Według niektórych autorów wskazane jest także poradnictwo genetyczne przy planowaniu prokreacji 5 .
Leczenie
Ogólnie przyjęte protokoły dotyczące leczenia TB nie zostały dotychczas opracowane. W praktyce należy rozważyć leczenie radykalne, farmakologiczne lub połączenie tych metod 18 . Należy podkreślić, że metoda leczenia powinna być adekwatna do grubości i głębokości naciekania guza, który z racji swego pochodzenia mieszkowego jest zlokalizowany w środkowych warstwach skóry. W odniesieniu do farmakoterapii ważne jest, by uwzględniać poznane mechanizmy patogenetyczne.
Leczenie zabiegowe
W przypadku sporadycznych TB, niezależnie od utkania histologicznego, najlepszą opcją jest zabieg chirurgiczny. Jako guzy łagodne TB nie wymagają szerokiego marginesu wycięcia, chyba że są przesłanki do podejrzenia zezłośliwienia. Chirurgia mikrograficzna Mohsa może być zastosowana w przypadku plaque-type TB 3 .
Również w odniesieniu do TB wyrastających na podłożu znamienia łojowego wycięcie chirurgiczne jest najlepszą opcją terapeutyczną. Zaleca się, o ile to możliwe, wycięcie całego znamienia łojowego wraz z guzem wtórnym u osób dorosłych lub wcześniej, w okresie okołopokwitaniowym, aby zapobiec rozwojowi TB i innych guzów wtórnych.
Dużym problemem jest leczenie licznych TB w przebiegu CCS. W przypadkach zmian drobnych i mnogich metody alternatywne, takie jak elektrochirurgia, łyżeczkowanie z elektrokoagulacją podstawy, waporyzacja laserowa (lasery ablacyjne CO2, Er:YAG, laser diodowy) lub kriochirurgia, także znajdują zastosowanie, chociaż z powodu podobieństwa klinicznego do BCC i innych łagodnych bądź złośliwych nowotworów wskazane jest potwierdzenie rozpoznania badaniem histopatologicznym. Nie jest ono możliwe w przypadku wspomnianych metod radykalnego zniszczenia guza 19, 20 .
Chociaż w literaturze są dostępne doniesienia o pozytywnych efektach stosowania peelingów chemicznych i dermabrazji u pacjentów z BSS/MFT/CCS, należy zwrócić uwagę, że metody te działają powierzchownie, dają raczej niewielkie i krótkotrwałe efekty kosmetyczne w przypadku drobnych, powierzchownych zmian, nie zapobiegają nowym zmianom i wiążą się z dużą częstością nawrotów, a ich stosowanie nie ma uzasadnienia patogenetycznego 21, 22 .
Farmakoterapia
Leczenie radykalne nie eliminuje ryzyka nawrotu oraz nie zapobiega pojawieniu się nowych zmian. Aktualnie dostępne w literaturze dane dotyczące leczenia farmakologicznego omawianych patologii z uwzględnieniem poznanych mechanizmów patofizjologicznych, choć są dosyć ograniczone i dotyczą stosunkowo krótkich okresów obserwacji, można uznać za obiecujące 20 .
Lepsza znajomość mechanizmów patofizjologicznych białka CYLD, w szczególności dotyczących jego działania hamującego wobec czynnika jądrowego κB (NF-κB – nuclear factor κB) indukowanego przez czynnik martwicy nowotworów (TNF – tumor necrosis factor), może prowadzić do rozważenia opcji terapeutycznych, takich jak antagoniści NF-κB (kwas acetylosalicylowy lub niesteroidowe leki przeciwzapalne) i anty-TNF-α (adalimumab). Aktywacja szlaku mTOR (mammalian target of rapamycin) w patogenezie TE uzasadnia zastosowanie inhibitora tego szlaku – sirolimusu, a potwierdzenie roli mutacji genu PTCH1 odpowiadającego za aktywację szlaku Hedgehog w MFT2 zachęca do wykorzystania wismodegibu w leczeniu farmakologicznym TB.
Do tej pory żaden lek nie został zarejestrowany w terapii łagodnych zmian w przebiegu CCS. We wszystkich opisywanych przypadkach leczenia farmakologicznego odsetek odpowiedzi określono na podstawie obserwacji klinicznej i/lub subiektywnej oceny pacjenta, stwierdzając brak odpowiedzi lub częściową odpowiedź. Całkowitej odpowiedzi nie udało się uzyskać u żadnego z opisanych pacjentów bez względu na metodę farmakoterapii. Działania niepożądane związane z leczeniem farmakologicznym były na ogół dobrze tolerowane i nie stanowiły powodu przerwania terapii.
Sirolimus (rapamycyna)
Jest to wybiórczy lek immunosupresyjny, który wiąże się ze specyficznym białkiem cytozolowym FKPB-12, a powstający kompleks FKPB-12–sirolimus hamuje aktywację mTOR, co prowadzi do zablokowania niektórych swoistych szlaków przewodzenia sygnałów w komórce. Identyfikacja szlaku mTOR w patogenezie TE odsłoniła nowe możliwości terapeutyczne dla pacjentów z CCS. W Polsce sirolimus jest dostępny w postaci roztworu doustnego oraz tabletek drażowanych, a wskazania rejestracyjne są ograniczone do zapobiegania odrzuceniu przeszczepu nerki oraz leczenia sporadycznej limfangioleiomiomatozy. W Japonii sirolimus w postaci 0,2% żelu został zarejestrowany w leczeniu TSC.
W literaturze opisano efekty stosowania sirolimusu miejscowo (w postaci kremu w stężeniu 0,2-1% aplikowanego 2 × dziennie przez 5-12 miesięcy) u kilkorga dzieci i dorosłych z mnogimi TE. Choć terapia nie doprowadziła do całkowitego ustąpienia zmian u żadnej z leczonych osób, to u większości spowodowała częściowe spłaszczenie guzków. Zapobiegała także powstawaniu nowych zmian u pacjentów obciążonych mutacją genu CYLD oraz u osób z mnogimi TE bez potwierdzenia tej mutacji. Sirolimus może zatem zatrzymać szybki rozrost mnogich TE, zwłaszcza jeśli zostanie wcześnie wdrożony u dzieci z ograniczoną liczbą zmian i niewielkim nasileniem choroby, co może zapobiec konieczności wielokrotnych operacji i zminimalizować psychospołeczne obciążenie chorobą. Leczenie miejscowe sirolimusem może być zoptymalizowane dzięki zastosowaniu metod poprawiających penetrację leku, takich jak okluzja, współczesne formulacje farmaceutyczne, wcześniejsze stosowanie laserów ablacyjnych 20, 21, 22, 23, 24, 25 .
Imikwimod
Jest modulatorem odpowiedzi immunologicznej. Wykazuje działanie przeciwnowotworowe i przeciwwirusowe. Jest agonistą receptorów Toll-podobnych i stymuluje wydzielanie m.in. TNF-α oraz IFN-α poprzez aktywację NF-κB 26 .
W Polsce imikwimod jest dostępny w postaci 5% kremu do stosowania zewnętrznego. Został zarejestrowany w leczeniu kłykcin kończystych zewnętrznych narządów płciowych i okolic odbytu, powierzchownych ognisk BCC oraz rogowacenia słonecznego bez cech przerostu i hiperkeratozy, o typowym przebiegu klinicznym, na twarzy lub owłosionej skórze głowy u chorych z wydolnym układem immunologicznym, gdy inne metody leczenia miejscowego są przeciwwskazane albo nieodpowiednie.
W literaturze można znaleźć prace kazuistyczne dotyczące zastosowania 5% imikwimodu w kremie (w monoterapii) u pacjentów z mnogimi TE w przebiegu BSS (3-7 × w tygodniu przez 3-8 miesięcy), z widoczną przynajmniej częściową poprawą. Obiecujące wyniki uzyskali także badacze, którzy wdrożyli u 11-letniej dziewczynki terapię skojarzoną 5% imikwimodem w kremie z 1% żelem z tretynoiną – w ciągu 3 lat ustąpiło 80% zmian. Inny rodzaj terapii złożonej zastosowano u pacjenta z MFT (ablacja laserem CO2, a następnie aplikacja 5% imikwimodu w kremie 5 × w tygodniu) 20 .
Wismodegib
Jest drobnocząsteczkowym inhibitorem szlaku Hedgehog. Przekazywanie sygnałów tym szlakiem przez białko SMO (smoothened transmembrane protein) prowadzi do aktywacji i lokalizacji w jądrze komórkowym czynników transkrypcyjnych GLI (glioma-associated oncogene) oraz indukcji docelowych genów szlaku Hedgehog. Wiele z tych genów odgrywa rolę w proliferacji, przeżyciu i różnicowaniu komórek. Wismodegib wiąże się z białkiem SMO i hamuje jego funkcję, prowadząc do zablokowania transdukcji sygnału przekazywanego szlakiem Hedgehog.
W Polsce wismodegib jest dostępny w postaci kapsułek twardych, zawierających 150 mg preparatu. Został zarejestrowany w leczeniu BCC objawowego z przerzutami oraz miejscowo zaawansowanego BCC bez możliwości leczenia chirurgicznego lub radioterapii.
Wismodegib p.o. zastosowano u pacjenta z mnogimi TE, u którego wskazaniem do podania leku były równocześnie występujące zaawansowany i przerzutowy BCC oraz złośliwy rak trichoblastyczny. Po 2 miesiącach leczenia zaobserwowano znaczną regresję guzków na twarzy, przy braku odpowiedzi ze strony guzów złośliwych. Znaczącą poprawę uzyskano także u innego pacjenta z mnogimi TE, stosując wismodegib doustnie w połączeniu z ablacją laserem CO2 licznych guzków na twarzy 20 .
Adalimumab
Jest to rekombinowane ludzkie przeciwciało monoklonalne, które wiąże się swoiście z TNF-α i hamuje jego aktywność poprzez blokowanie wiązania z receptorami TNFR1 (p55) i TNFR2 (p75) na powierzchni komórki. W Polsce adalimumab jest wskazany w leczeniu niektórych postaci reumatoidalnego zapalenia stawów, czynnego wielostawowego młodzieńczego idiopatycznego zapalenia stawów, zapalenia stawów z towarzyszącym zapaleniem przyczepów ścięgnistych, łuszczycy zwykłej i łuszczycowego zapalenia stawów, zesztywniającego zapalenia stawów kręgosłupa, osiowej spondyloartropatii bez zmian radiograficznych charakterystycznych dla zesztywniającego zapalenia stawów kręgosłupa, choroby Leśniowskiego-Crohna, wrzodziejącego zapalenia jelita grubego, ropnego zapalenia apokrynowych gruczołów potowych, nieinfekcyjnego zapalenia błony naczyniowej oka. W naszym kraju obecnie są dostępne preparaty siedmiu producentów, a wskazania rejestracyjne do ich stosowania wg ChPL poszczególnych preparatów mogą się różnić.
Środki farmakologiczne, które mają działanie hamujące na funkcje NF-κB, mogą być potencjalnie przydatne w leczeniu MFT1. Fisher i Geronemus w 2010 r. opisali przypadek MFT o ciężkim przebiegu u 41-letniej kobiety, u której występowały nawroty zmian mimo poddania się licznym zabiegom laserowym w ciągu 15 lat. Osiągnięto bardzo wyraźną poprawę po leczeniu obejmującym kwas acetylosalicylowy podawany doustnie i adalimumab w iniekcjach podskórnych przez 8 miesięcy. Po leczeniu stwierdzono, że resztkowe TE były mniejsze i bardziej płaskie, a skóra pacjentki stała się mniej stwardniała. Zaobserwowano także znaczną poprawę w zakresie TE zlokalizowanych w okolicach brwi i przedniej linii włosów, czyli dwóch obszarach, które wcześniej nie były poddawane zabiegom ablacji laserowej, zatem poprawa stanowiła jedynie efekt farmakoterapii. W opisanym przypadku wpływ każdego z leków na wynik leczenia wydaje się jednak trudny do precyzyjnego określenia 20 .
Glikokortykosteroidy i niesteroidowe leki przeciwzapalne (jako czynniki hamujące szlak NF-κB)
W 2016 r. Mulder i wsp. opisali terapię cylindroma z wykorzystaniem kolejno serii doogniskowych iniekcji acetonidu triamcynolonu, diklofenaku w żelu stosowanego miejscowo przez 3 miesiące oraz serii doogniskowych wstrzyknięć kwasu salicylowego. Guzy leczone w taki sposób pozostały jednak niezmienione 27 . Podobnie niezadowalające wyniki uzyskano, stosując miejscowo kwas salicylowy w różnych stężeniach w leczeniu CCS, prawdopodobnie dlatego że jest on raczej słabym i nieswoistym blokerem aktywacji NF-κB 28 .
Retinoidy
Próby leczenia ogólnego mnogich TE retinoidami: izotretynoiną i acytretyną również nie przyniosły zachęcających wyników 21, 29 .
Nadtlenek benzoilu
Jest związkiem utleniającym, który działa miejscowo przeciwzapalnie, przeciwbakteryjnie, przeciwłojotokowo, łagodnie keratolitycznie, wysuszająco, miejscowo znieczulająco i przeciwświądowo, pobudza ziarninowanie oraz syntezę kolagenu. W Polsce jest dostępny w postaci 5% i 10% żelu. Może być stosowany miejscowo samodzielnie lub jako składnik złożonych preparatów przeciwtrądzikowych.
Okamura i wsp. opisali efekt stosowania nadtlenku benzoilu u 18-letniej kobiety z MFT. Po 3 miesiącach monoterapii zaobserwowali spłaszczenie i zmniejszenie grudek na twarzy oraz dalszą poprawę 7 miesięcy później. Nie wystąpiły żadne zdarzenia niepożądane. Zmiany skórne nigdy nie uległy zaostrzeniu i pozostawały pod dobrą kontrolą kliniczną przez co najmniej 2,5 roku obserwacji. Zdaniem autorów poprawę po zastosowaniu nadtlenku benzoilu u pacjentów z MFT można wytłumaczyć m.in. działaniem przeciwzapalnym poprzez hamowanie uwalnianych cytokin prozapalnych, w tym TNF-α 30 .
Pegkantratynib
W badaniu klinicznym II fazy, które obejmowało pacjentów z CCS (fenotypowym lub potwierdzonym w badaniu genetycznym), nie uzyskano oczekiwanej redukcji rozmiaru guzów leczonych miejscowo preparatem zawierającym pegkantratynib (inhibitor kinazy receptora tropomiozyny) w stężeniu 0,5%. Zaobserwowano redukcję bolesności guzów, potwierdzono bezpieczeństwo i dobrą tolerancję leczenia, ale stężenie leku uznano za niewystarczające, aby znieść sygnalizację kinazy receptora tropomiozyny 5, 31 .
Inne metody łączone
Opisano efekty dwóch kombinacji różnych metod (ablacyjny laser frakcjonowany Er:YAG i terapia fotodynamiczna [PDT – photodynamic therapy] z kwasem 5-aminolewulinowym; ablacyjny laser frakcjonowany Er:YAG i miejscowy imikwimod) w porównaniu z samym resurfacingiem lub samą PDT u 49-letniego mężczyzny rasy kaukaskiej z BSS, z 29-letnim wywiadem zmian skórnych na twarzy, dotychczas nieleczonego z tego powodu. Według zespołu badaczy połączenie serii zabiegów z użyciem ablacyjnego lasera frakcjonowanego Er:YAG i PDT pozwoliło uzyskać nieco lepszy wynik niż seria zabiegów resurfacingu w połączeniu z miejscowym imikwimodem, sam cykl resurfacingu lub sama seria PDT, a poprawa stanu skóry utrzymywała się 10 miesięcy po zakończeniu leczenia 32 .
Podsumowanie
Podstawą rozpoznania TB pozostaje badanie kliniczne i histopatologiczne.
Ogólnie przyjęte protokoły leczenia TB nie zostały dotychczas opracowane. W praktyce należy rozważyć leczenie radykalne, zachowawcze lub połączenie tych metod.
W przypadku zmian pojedynczych preferowaną metodą jest usunięcie chirurgiczne oraz badanie histopatologiczne w celu potwierdzenia rozpoznania i dokonania oceny doszczętności zabiegu. Jednak w niektórych sytuacjach dopuszcza się postawę wyczekującą pod warunkiem zapewnienia bacznej obserwacji.
W przypadku zmian mnogich wymagane jest wnikliwe różnicowanie, a postępowanie diagnostyczne powinno opierać się na badaniu klinicznym i histopatologicznym. Diagnostyka genetyczna jest uzasadniona i powinna wkrótce stać się standardem. Określenie wariantu patogenicznego w genie CYLD u pacjenta dotkniętego CCS oraz stwierdzenie innych mutacji wpływa nie tylko na poradnictwo genetyczne i decyzje dotyczące planowania rodziny, lecz także może być przydatne przy wyborze leczenia celowanego. Dobór metody leczniczej u takich pacjentów jest uzależniony od liczby, lokalizacji, rozmiaru zmian, podejrzenia zezłośliwienia, chorób współistniejących, aktualnie stosowanych leków, potencjalnych działań niepożądanych (np. łysienie w przypadku przyjmowania wismodegibu u ok. 25% osób), wieku, planów prokreacyjnych i docelowego efektu estetycznego oraz dostępności poszczególnych preparatów i metod niefarmakologicznych, w tym możliwości ekonomicznych. Uzasadnione wydaje się stosowanie terapii złożonej z leczenia zabiegowego w połączeniu z wczesnym i przewlekłym leczeniem farmakologicznym, chociaż w odniesieniu do tego ostatniego aktualnie dostępne dane naukowe pozostają bardzo ograniczone, opisywane metody terapii są słabo wystandaryzowane, a wszystkie preparaty były podawane off-label.
Należy mieć nadzieję, że zostaną opracowane skuteczne metody farmakologicznej profilaktyki zmian skórnych u osób z potwierdzonym genetycznie CCS.
U wszystkich pacjentów z CCS, którzy zgłosili zmiany w istniejących guzach, takie jak szybki wzrost, modyfikacja wyglądu w stosunku do innych, już istniejących zmian, krwawienia lub owrzodzenia guzów, zalecana jest pilna diagnostyka w celu ustalenia wskazań do wycięcia i poszerzonej diagnostyki obrazowej. Należy również badać okresowo gruczoły ślinowe. U pacjentów powyżej 40 r.ż. zgłaszających zaburzenia oddychania powinno się wykonać badanie radiologiczne płuc ze względu na potencjalne ryzyko wystąpienia cylindroma płuc. U wszystkich chorych zaleca się fotoprotekcję i unikanie radioterapii.
Abstract
The types, diagnosis and treatment of trichoblastoma
Trichoblastoma (TB) is a rare, benign tumor of hair origin, which most often occurs sporadically in the form of single, asymptomatic, dome-shaped and flesh-colored nodules or porcelain discs (desmoplastic type) on intact skin or within a sebaceous nevus. In its multiple form, it occurs as an element of the genetically determined CYLD cutaneous syndrome. TB most often affects the face. The risk of malignancy is low. Dermatoscopic examination may be helpful in the diagnosis of TB, but the final diagnosis is made based on histopathological examination results. The preferred treatment method for both single and multiple lesions is surgical excision. However, in relation to multiple TB in genetic syndromes, new hope has been given to patients thanks to new pharmacological agents based on recently discovered pathogenetic mechanisms, such as sirolimus, vismodegib, imiquimod or adalimumab, as a method to prevent new tumors or as a complementary therapy to surgical treatment.
- 1. Kutzner H, Kaddu S, Kanitakis J, et al. Trichoblastoma. In: WHO Classification of Skin Tumors. 4-th edition. Edited by Elder DE, Massi D, Scolyer RA, et al. Lyon: International Agency for Research on Cancer, 2018
- 2. Kazakov DV, Michal M, Kacerovska D, et al. Cutaneous Adnexal Tumors. Philadelphia: Volters Kluver Health, Lippinkott Williams & Wilkins, 2012
- 3. Woods AD, Grushchakc S, Rakita U, et al. Plaque variant trichoblastoma – An unusually aggressive neoplasm: Presentation of 11 cases in 4 individuals and a review of the literature. JAAD Case Reports 2023;(36):108-12
- 4. Nagy N, Dubois A, Szell M, et al. Genetic Testing in CYLD Cutaneous Syndrome: An Update. Appl Clin Genet 2021;14:427-44
- 5. Dubois A, Rajan N. CYLD Cutaneous Syndrome. 2020 Apr 16. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle, 1993-2023
- 6. Borzęcki A, Szponar-Bojda A, Szubstarski F. Trichoepithelioma – trudności diagnostyczne. Dermatol Rev/Przegl Dermatol 2013;100(2):110-7
- 7. Harada H, Hashimoto K, Ko MSH. The Gene for Multiple Familial Trichoepithelioma Maps to Chromosome 9p21. J Invest Dermatol 1996;107(1):41-3
- 8. Amaaoune F, Zidane W, Aksim M, et al. Multiple non-familial trichoepitheliomas: A rare case and a review of the literature. Our Dermatology Online/Nasza Dermatologia Online 2023;14(3):307-10
- 9. Sood S, Gupta M, Sharma RK, et al. Multiple Non-Familial Trichoepitheliomas in a Nine Year Child. Nepal J Dermatol Venereol Leprol 2019;17(1):76-8
- 10. Liu Y, Banka S, Huang Y, et al. Germline intergenic duplications at Xq26.1 underlie Bazex-Dupré-Christol basal cell carcinoma susceptibility syndrome. Br J Dermatol 2022;187(6):948-61
- 11. Sikorska M, Szczerkowska-Dobosz A, Purzycka-Bohdan D, et al. Włosy skręcone i mnogie prosaki twarzy jako wyraz dysplazji ektodermalnej u bliźniaczek monozygotycznych. Dermatol Rev/Przegl Dermatol 2014;101(1):35-9
- 12. Pitarch G, Botella-Estrada R. Dermiscopic Findings in Trichoblastoma. Actas Dermosifiliogr 2015;106(9):45-8
- 13. Sławińska M, Płaszczyńska A, Lakomy J, et al. Significance of Dermoscopy in Association with Clinical Features in Differentiation of Basal Cell Carcinoma and Benign Trichoblastic Tumours. Cancers 2022;14(16):3964
- 14. Kitakamura S, Hata H, Imafuku K, et al. Basal Cell Carcinoma or Trichoblastoma Dermoscopic Examination of Black Macules Developing in the Same Nevus Sebaceus. Case Rep Oncol 2016;9(1):143-7
- 15. Pampena R, Peccerillo F, Marghoob NG, et al. Peritumoural clefting as a key feature in differentiating basal cell carcinoma from trichoblastoma through in vivo reflectance confocal microscopy. J Eur Acad Dermatol Venereol 2019;33(5):e201-3
- 16. Pogorzelska-Antkowiak A, Antkowiak R, Poloczek R. Trichoblastoma in reflectance confocal microscopy. Australas J Dermatol 2023;64(1):e89-90
- 17. Cazzato G, Cimmino A, Colagrande A, et al. The Multiple Faces of Nodular Trichoblastoma: Review of the Literature with Case Presentation. Dermatopathology 2021;8(3):265-70
- 18. Dissanayaka DWVN, Dassanayaka DKB, Jayasooriya PR. Clinical, Histopathological, and Management Challenges of Multiple Familial Trichoepithelioma: A Case Report of a Patient Presenting with Multiple Facial Papules. Case Rep Dent 2020;2020:5648647
- 19. Chaudhary S, Dayal S. Radiofrequency ablation: a safe and economical modality in treatment of Brooke-Spiegler syndrome. Dermatol Online J 2012;18(8):7
- 20. Lobo Y, Blake T, Wheller L. Management of multiple trichoepithelioma: A review of pharmacological therapies. Australas J Dermatol 2021;62(2):e192-200
- 21. du Toit JP, Schneider JW, Visser WI, et al. The clinicopathological spectrum of trichoepitheliomas: a retrospective descriptive study. Int J Dermatol 2016;55(3):270-7
- 22. Latif S, ul Bari A. Multiple familial trichoepitheliomas: A rare autosomal dominant skin tumor. J Pak Assoc Dermatol 2021;31(3):546-9
- 23. Dreyfus I, Onnis G, Tournier E, et al. Effect of Topical Rapamycin 1% on Multiple Trichoepitheliomas. Acta Derm Venereol 2019;99(4):454-55
- 24. Shimizu A, Toyoda A, Motegi S-I, et al. First Japanese case of trichoepithelioma papulosum multiplex successfully treated with sirolimus gel. J Dermatol 2020;47(5):e197-8
- 25. Capusan TM, Noguera-Morel L, Bueno-Martínez E, et al. Multiple familial trichoepitheliomas: Ultrasonographic findings. Skin Res Technol 2020;26(1):137-9
- 26. Omran FAE, Nofal A, Nofal H. An Insight about Possible Role of Imiquimod in Dermatology and Possible Benefit on Plane Warts: Review Article. Egypt J Hosp Med 2023;91:4246-50
- 27. Mulder KM, Crijns MB, Doorn R. Efficacy of intralesional corticosteroids in the treatment of cylindromas. Clin Exp Dermatol 2016;41(5):578-80
- 28. Oosterkamp HM, Neering H, Nijman SMB, et al. An evaluation of the efficacy of topical application of salicylic acid for the treatment of familial cylindromatosis. Br J Dermatol 2006;155(1):182-5
- 29. Van Voorst Vader PC, Van Oostveen F, Hasper MF. Trichoepithelioma, cystic acne and 13-cis-retinoic acid. Acta Derm Venereol 1984;64(4):360-1
- 30. Okamura S, Oyama N, Hasegawa M. The First Case Report of Multiple Familial Trichoepitheliomas Responding Successfully to Topical Benzoyl Peroxide: A Possible Therapeutic Action Underlying Structural Turnover and Antiinflammation. Indian J Dermatol 2022;67(1):67-8
- 31. Danilenko M, Stamp E, Stocken DD, et al. Targeting Tropomyosin Receptor Kinase in Cutaneous CYLD Defective Tumors With Pegcantratinib: The TRAC Randomized Clinical Trial. JAMA Dermatol 2018;154(8):913-21
- 32. Lopiccolo MC, Sage RJ, Kouba DJ. Comparing Ablative Fractionated Resurfacing, Photodynamic Therapy, and Topical Imiquimod in the Treatment of Trichoblastomas of Brooke-Spiegler Syndrome: A Case Study. Dermatol Surg 2011;37(7):1047-50
Następny artykuł:

 Dodaj do ulubionych
Dodaj do ulubionych
