



Co znajdziesz w artykule?
- Arytmogenna kardiomiopatia prawokomorowa – schorzenie związane z funkcjonalnymi lub strukturalnymi nieprawidłowościami mięśnia sercowego
- Trudności i wyzwania w ustaleniu ostatecznego rozpoznania
- Aktualna wiedza na temat możliwości diagnostycznych, z uwzględnieniem najnowszych zaleceń towarzystw kardiologicznych i stanowisk grup ekspertów
Spis treści
Arytmogenna kardiomiopatia prawokomorowa (ARVC – arrhythmogenic right ventricular cardiomyopathy) zalicza się do grupy schorzeń związanych z funkcjonalnymi lub strukturalnymi nieprawidłowościami mięśnia sercowego 1 . Pierwsze doniesienia o „nowym” rodzaju kardiomiopatii pochodzą z lat 80. XX w. Wówczas to Guy Fontaine oraz Frank Marcus opisywali przebudowę włóknisto-tłuszczową wolnej ściany prawej komory z towarzyszącym zanikiem kardiomiocytów, co nasuwało podejrzenie dysplazji miokardium
2, 3 . Pojawiające się nieprawidłowości architektury ścian prowadziły do zaburzenia kurczliwości mięśnia sercowego ze stopniową rozstrzenią komór oraz powstania blizn będących substratem dla arytmii komorowej 4 .
Mimo upływu czasu i dokładniejszego poznania patofizjologii nadal istnieją liczne wątpliwości nie tylko w wyborze optymalnej terapii, lecz także w ustaleniu ostatecznego rozpoznania ARVC. Przyczynia się do tego przewlekły charakter procesu chorobowego, który przekłada się na rozbieżność w interpretacji wyników seryjnie wykonywanych badań diagnostycznych. To z kolei utrudnia analizy kohortowe chorych oraz ustalenie optymalnej terapii schorzenia w zależności od stopnia zaawansowania. Wykładnikiem tej sytuacji są częste aktualizacje zaleceń dotyczących rozpoznawania i leczenia chorych z ARVC 5, 6, 7 .
Celem niniejszej pracy jest przedstawienie aktualnej wiedzy w zakresie patofizjologii, najczęstszych manifestacji schorzenia oraz możliwości diagnostycznych pozwalających ustalić ostateczne rozpoznanie ARVC.
Epidemiologia
Rzeczywista częstość występowania ARVC nie jest dokładnie poznana. Wynika to nie tylko z trudności w postawieniu ostatecznego rozpoznania wśród chorych żyjących, lecz także z niedostatecznej diagnostyki (w tym genetycznej) u ofiar nagłej arytmicznej śmierci sercowej. Na podstawie danych ekstrapolowanych z dużych analiz częstość występowania ARVC szacuje się na 1:1300 osób 8 . Ryzyko to jest istotnie wyższe u krewnych pierwszego stopnia, zwłaszcza u rodzeństwa, w przypadku którego schorzenie występuje trzykrotnie częściej niż u dzieci i rodziców 9 . Płeć męska wiąże się z częstszym występowaniem oraz cięższym przebiegiem klinicznym schorzenia. Związek ten tłumaczy się wpływem testosteronu na ekspresję wadliwych genów oraz większą intensywnością podejmowanego wysiłku fizycznego 4 . U pacjentów uprawiających sporty wytrzymałościowe objawy niewydolności serca oraz groźne arytmie komorowe pojawiają się w młodszym wieku 10 .
Przebieg naturalny ARVC
ARVC jest najczęściej rozpoznawana między drugą a piątą dekadą życia 9 . W dzieciństwie choroba manifestuje się wyjątkowo rzadko (na tym etapie życia diagnoza jest stawiana u 15% chorych) 8 . Typowo, w naturalnym przebiegu schorzenia, wyróżnia się cztery etapy. W trakcie pierwszego nie stwierdza się makroskopowych zmian w obrębie miokardium. Nie wyklucza to jednak ryzyka występowania epizodów arytmicznych, w tym prowadzących do nagłego zgonu sercowego (SCD – sudden cardiac death) 4, 11 . Z czasem następuje okres jawny klinicznie ze zmianami funkcjonalnymi oraz strukturalnymi mięśnia sercowego. Pacjenci w tej fazie choroby mogą odczuwać kołatania serca, niespecyficzne dolegliwości bólowe w klatce piersiowej oraz duszności; w skrajnych przypadkach mogą również występować omdlenia 12, 13 . Fazie trzeciej towarzyszy postępująca dysfunkcja prawej, a z czasem także lewej komory. W schyłkowym stadium choroby dochodzi do rozstrzeni obu komór serca z upośledzeniem ich kurczliwości. Obraz ten może nasuwać mylne podejrzenie kardiomiopatii rozstrzeniowej 4 . To na tym etapie ryzyko wystąpienia SCD jest najwyższe – rocznie sięga 4% 14, 15 . Główną przyczyną jest nasilenie objawów niewydolności serca (59%), co również zwiększa ryzyko wystąpienia złożonych arytmii komorowych (29%) 12 .
Sugeruje się, że stabilne fazy choroby są zaburzane okresami zaostrzenia pod postacią zapalenia mięśnia sercowego (tzw. hot phase). Nie wiadomo, czy są one reakcją organizmu na uszkodzenie mięśnia sercowego wynikające z kardiomiopatii, czy też powodują je czynniki zewnętrzne, takie jak wirusy kardiotropowe 4 . Fazy zaostrzenia choroby (hot phase) mogą imitować ostry zespół wieńcowy, ze zmianami zapisu EKG w zakresie odcinka ST i samych załamków T oraz uwolnieniem biomarkerów wskaźnikowych martwicy mięśnia sercowego 4 .
Patofizjologia i tło genetyczne
Podstawą patofizjologiczną rozwijającego się schorzenia jest postępująca dysfunkcja złącza między kardiomiocytami (connexon), na które składają się desmosomy, połączenia szczelinowe, adherencyjne oraz kanały jonowe 16 . Uszkodzenia, do których dochodzi podczas progresji choroby, prowadzą do utraty połączeń międzykomórkowych, co skutkuje apoptozą kardiomiocytów. Konsekwencją wspomnianego zjawiska są zaburzenia przezkanałowego przepływu jonów oraz różnicowania komórek progenitorowych, skutkujące włóknisto-tłuszczowym remodelingiem mięśnia sercowego 4, 16 . Proces ten w pierwszej kolejności postępuje w warstwie epikardialnej i szerzy się do endokardium, zajmując w końcowym stadium całą grubość ściany, co prowadzi do jej ścieńczenia i powstawania tętniaków 4 . W rzadkich przypadkach uszkodzenie może również dotyczyć połączeń międzykomórkowych w obrębie skóry i dawać obraz zespołu Naxos, cechujący się oprócz zajęcia miokardium występowaniem hiperkeratozy oraz zaburzeń wzrostu owłosienia głowy (tzw. wełnistych włosów) 17 . Za większość przypadków choroby odpowiada 8 mutacji genów, głównie o dziedziczeniu autosomalnym dominującym (5 genów desmosomalnych – PKP2, DSP, DSG2, DSC2 i JUP – oraz 3 geny niedesmosomalne – TMEM43, DES, PLN) 16, 18 . U 4-16% pacjentów współwystępuje kilka patogennych mutacji. Mogą one dotyczyć tego samego genu (heterozygoty złożone) lub różnych genów (heterozygoty digeniczne), a ich współistnienie wiąże się ze zwiększonym ryzykiem wystąpienia arytmii oraz SCD 4 . Mimo analizy genetycznej u prawie 40% chorych nie udaje się znaleźć patogennej mutacji 19 . Wśród tych pacjentów objawy chorobowe i arytmie komorowe występują później niż u chorych z ustaloną znaną mutacją genetyczną 20 .
Badania diagnostyczne
Spoczynkowy zapis EKG
Nieprawidłowości w zapisie spoczynkowego elektrokardiogramu występują u większości pacjentów z ARVC (81-88%) 9, 21 . Zmiany te pojawiają się wraz z progresją schorzenia, a stopień ich nasilenia może mieć charakter dynamiczny w kolejnych zapisach 22, 23 . W przypadku rozpoznania postawionego na podstawie innych metod diagnostycznych zapis EKG pozostaje prawidłowy u ponad połowy chorych 14 . Z kolei po 6 latach od wystąpienia pierwszego epizodu arytmii komorowej u każdego pacjenta pojawiają się nieprawidłowości w zapisie EKG 24 .
Wśród odchyleń spoczynkowego elektrokardiogramu stwierdzanych u chorych z ARVC wymienia się:
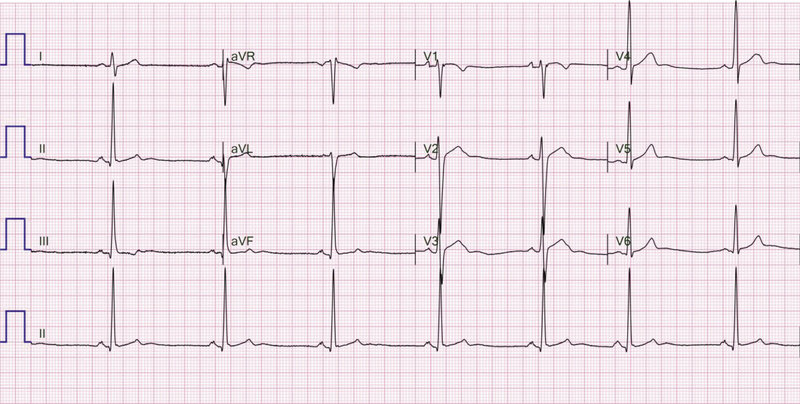
Rycina 1. Zapis 12-kanałowego EKG 36-letniego sportowca ze zdiagnozowaną arytmogenną kardiomiopatią prawokomorową, u którego w rezonansie magnetycznym serca stwierdzono regionalne zaburzenia kurczliwości prawej komory. W zapisie widoczne TAD (terminal activation delay) 63 ms w odprowadzeniach V1 i V2
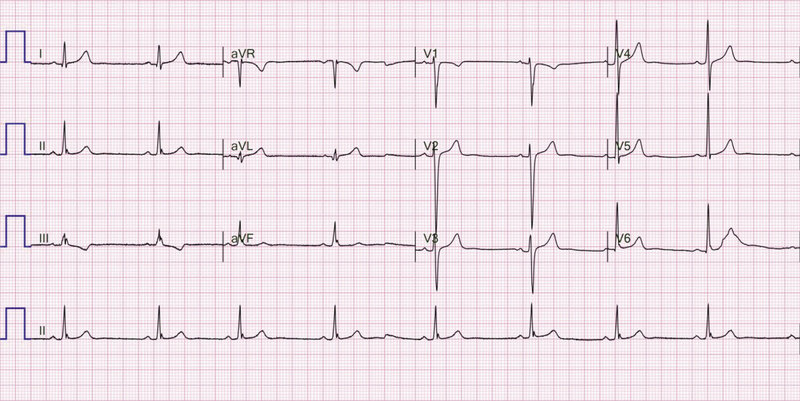
Rycina 2. Zapis 12-odprowadzeniowego EKG 22-letniego pacjenta z arytmogenną kardiomiopatią prawokomorową po przebytym nagłym zatrzymaniu krążenia w mechanizmie migotania komór. Widoczna fragmentacja zespołu QRS w odprowadzeniach znad ściany dolnej. Ponadto iloraz sumy czasu trwania zespołów QRS w odprowadzeniach V1-V3 oraz czasu trwania zespołu QRS w odprowadzeniach V4-V6 wynosi ok. 1,5
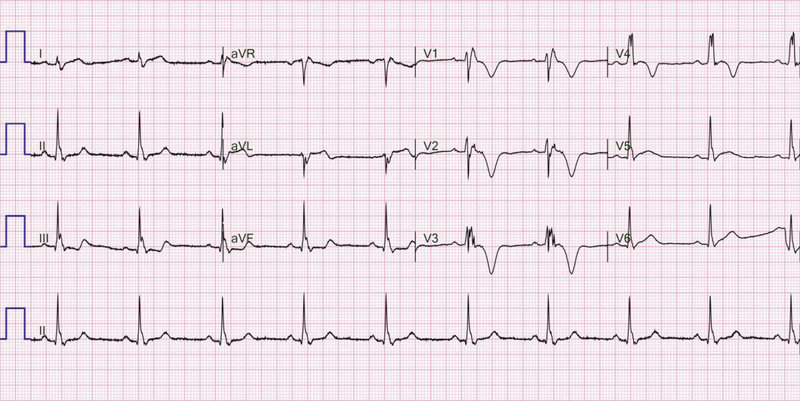
Rycina 3. Zapis 12-odprowadzeniowego EKG 29-letniego pacjenta z arytmogenną kardiomiopatią prawokomorową z towarzyszącym blokiem prawej odnogi pęczka Hisa. Iloraz amplitudy załamka r’/s jest mniejszy od jedności
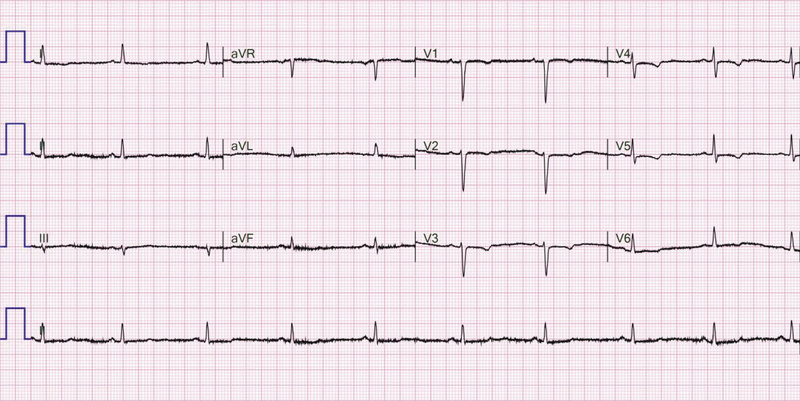
Rycina 4. Zapis 12-kanałowego EKG 36-letniej chorej z zaawansowaną postacią arytmogennej kardiomiopatii prawokomorowej z ujemnymi załamkami T w odprowadzeniach V1-V6
- Zmiany wynikające z zaburzenia procesu depolaryzacji prawej komory serca:
- Wydłużony czas narastania załamka S (liczony od nadiru załamka S do linii izoelektrycznej ≥55 ms w odprowadzeniach V1-V3) – zmiana opisywana u 19-95% pacjentów z ARVC 13, 25, 26, 27, 28, 29 . Dokładny pomiar końca załamka S może być zaburzony przez jego fragmentację lub pojawienie się fali epsilon. Z tego powodu został wprowadzony nowy, alternatywny pomiar liczony od nadiru załamka S do końca wszelkich wychyleń zespołu QRS (TAD – terminal activation delay) 28 . Występuje u pacjentów z podwyższonym ryzykiem arytmii komorowej 30 oraz rozstrzeni i dysfunkcji skurczowej prawej komory serca 31 . Cechuje go 59% czułość i 83% swoistość diagnostyczna 32 , jednak nie jest przydatny w przypadku występowania bloku prawej odnogi pęczka Hisa (RBBB – right bundle branch block) 33 . Przykład elektrokardiogramu z TAD ≥55 ms przedstawiono na rycinie 1.
- Fragmentacja zespołu QRS w odprowadzeniach V1-V3 – stwierdzana w 38-85% przypadków 25, 26, 34 . Obecność tej nieprawidłowości dotyczy pacjentów z wyższym ryzykiem występowania komorowych zaburzeń rytmu serca, a czułość i swoistość w ich prognozowaniu opisywano odpowiednio na poziomie 57% i 99,7% 34 . Przykład zapisu EKG ze wspomnianą zmianą przedstawiono na rycinie 2.
- Fala epsilon – definiowana jako niskoamplitudowy powtarzalny sygnał, dający obraz zawęźlenia na ramieniu wstępującym załamka S w odprowadzeniach V1-V3. Stwierdza się ją u 3-32% chorych z ARVC i w przeszłości była uważana za zmianę wysoce swoistą dla tego schorzenia 13, 25, 26, 27, 28, 29 . Aktualnie wiadomo, że może występować również w sarkoidozie, zawale prawej komory, anomalii Uhla, po chirurgicznej korekcji tetralogii Fallota, w zespole Brugadów oraz w przebiegu niedokrwistości sierpowatej 35 . Co więcej, jej znaczenie jest obecnie marginalne, ponieważ, jak wykazano, istnieją znaczne rozbieżności w interpretacji poszczególnych zmian EKG jako fali epsilon 36 .
- Poszerzenie zespołu QRS ≥110 ms w odprowadzeniach V1-V3 – zmiana opisywana u 18-75% badanych pacjentów z ARVC 13, 26, 27, 28, 29 . Było to jedno z oryginalnych kryteriów wskazanych przez Fontaina 37 . Wiele kontrowersji budził brak adnotacji, czy może być ono stosowane u pacjentów z zaburzeniami przewodnictwa śródkomorowego 33 . W 2003 r. zaproponowano wykorzystanie alternatywnego wskaźnika mającego cechować się wyższą czułością i swoistością diagnostyczną, tj. ilorazu sumy czasu trwania zespołów QRS w odprowadzeniach V1, V2, V3 oraz V4, V5, V6 ≥1,2 38 . Jednak ze względu na brak wartości klinicznej 33 parametr ten nigdy nie znalazł się w oficjalnych zaleceniach, a od 2010 r. czas trwania zespołów QRS w odprowadzeniach V1-V3 nie jest w ogóle wymieniany jako kryterium diagnostyczne. Zmianę uchwyconą w zapisie elektrokardiograficznym przedstawiono na rycinie 2.
- Częściowy blok przewodnictwa śródkomorowego – zdefiniowany przez Fontaina jako wydłużenie czasu trwania zespołu QRS w odprowadzeniach V1-V3 o co najmniej 25 ms od czasu trwania QRS w odprowadzeniu V6 33 . Występuje w 7-73% opisywanych przypadków ARVC 25, 27, 29 . Nie jest miarodajny u pacjentów z zaburzeniami przewodnictwa śródkomorowego i nigdy nie był wskazywany jako kryterium diagnostyczne 33 . Nie ma również wpływu na predykcję SCD 25 .
- Iloraz amplitudy załamka r’/s w odprowadzeniu V1 – parametr zaproponowany przez Jaina i wsp. dla chorych z RBBB. Wartość ilorazu <1 cechuje się 88% czułością oraz 86% swoistością w rozpoznawaniu ARVC, co sprawia, że jest to najrzetelniejszy parametr diagnostyczny w przypadku zaburzeń przewodnictwa śródkomorowego 33 . Przykład zapisu EKG ze zmianą przedstawiono na rycinie 3.
- Zmiany wynikające z zaburzenia procesu repolaryzacji prawej komory serca:
- Ujemny załamek T w odprowadzeniach V1-V3 (TWI – T wave inversion) – zmiana obecna w 10-87% przypadków ARVC 13, 25, 26, 27, 28, 29 . W związku z optymalnym stosunkiem czułości i swoistości diagnostycznej wydaje się najbardziej przydatnym elektrokardiograficznym kryterium diagnostycznym 33 . Pojawienie się TWI w większej liczbie odprowadzeń odzwierciedla dylatację prawej komory oraz zwiększa ryzyko wystąpienia arytmii komorowej 30, 31 , jednak jego izolowana obecność w V4-V6 jest mniej istotna 32 . Przykład zmiany w odprowadzeniach V1-V6 przedstawiono na rycinie 4.
- Niski woltaż zespołów QRS w odprowadzeniach kończynowych – po wykluczeniu alternatywnych przyczyn, takich jak sarkoidoza, otyłość, obecność płynu w worku osierdziowym, rozedma płuc, może świadczyć o ARVC. Stwierdzenie tej nieprawidłowości zapisu EKG jest skojarzone z obecnością włóknisto-tłuszczowej blizny zlokalizowanej subepikardialnie w obrębie ściany lewej komory 7 .
Zestawienie
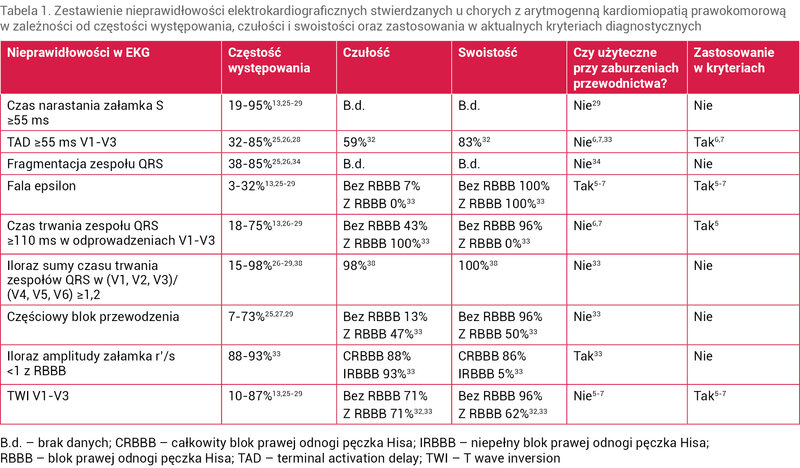
Tabela 1. Zestawienie nieprawidłowości elektrokardiograficznych stwierdzanych u chorych z arytmogenną kardiomiopatią prawokomorową w zależności od częstości występowania, czułości i swoistości oraz zastosowania w aktualnych kryteriach diagnostycznych
Zaburzenia rytmu serca w długoczasowej rejestracji EKG
Migotanie przedsionków jest najczęstszą tachyarytmią nadkomorową stwierdzaną u 9-30% chorych z ARVC 8 . Ryzyko wystąpienia tej arytmii jest wyższe niż w populacji ogólnej, a jej obecność istotnie obciąża rokowanie oraz świadczy o progresji dysplazji jam serca 39 . Na możliwość pierwotnego zajęcia przez proces chorobowy mięśniówki przedsionków zwrócili uwagę m.in. Zghaib i wsp. Pogorszenie funkcji tych jam serca zostało stwierdzone na podstawie badania rezonansu magnetycznego, co korelowało z następczym pojawieniem się arytmii przedsionkowych 40 .
Wśród pacjentów z ARVC komorowe zaburzenia rytmu mogą występować z różnym nasileniem, od pojedynczych dodatkowych pobudzeń komorowych do złożonych form arytmii prowadzących do SCD. Charakterystyczną morfologią dla ARVC są arytmie komorowe o morfologii bloku lewej odnogi pęczka Hisa (LBBB – left bundle branch block) oraz osi górnej, cechujące się 98% swoistością diagnostyczną 32 . Rejestracja w zapisie monitorowania EKG metodą Holtera ponad 500 dodatkowych pobudzeń komorowych w trakcie doby jest uznanym, czułym kryterium diagnostycznym, przydatnym w screeningu pacjentów z podejrzeniem choroby 32 . Pacjenci, u których nie rejestrowano arytmii komorowej, stanowią jedynie mały odsetek chorych i są obciążeni najmniejszym ryzykiem sercowo-naczyniowym 12 . Wyższe ryzyko wystąpienia utrwalonej arytmii komorowej jest związane zarówno z czynnikami sercowymi, jak i pozasercowymi. Częściej dotyczy to pacjentów objawowych 30 , z wyższą punktacją w skali diagnostycznej ARVC z 2010 r., z dysfunkcją skurczową lewej lub prawej komory serca (obniżona frakcja wyrzutowa lewej komory [LVEF – left ventricular ejection fraction] lub frakcja skracania powierzchni prawej komory [RV-FAC – right ventricular fractional area change]), podwyższonym stężeniem N-końcowego fragmentu propeptydu natriuretycznego typu B (NT-proBNP – N-terminal pro B-type natriuretic peptide) oraz wysokoczułej sercowej izoformy troponiny T (hs-cTnT – high-sensitive cardiac troponin T), co jest skorelowane z większym stopniem zaawansowania choroby 41 . Wśród czynników pozasercowych wymienia się m.in.: podwyższone stężenie testosteronu u chorych płci męskiej lub obniżone stężenie estradiolu u kobiet, w tym związane z okresem około- i pomenopauzalnym, co tłumaczy późniejszy początek objawów 41 .
SCD może być pierwszym objawem ARVC 13 . Arytmia komorowa, u której podstaw leży dysplazja miokardium, jest powodem 4-23% wszystkich SCD u młodych osób (<35 r.ż.) 42 i bywa opisywana jako ich główna przyczyna 43 . Typowo wynika ona z migotania komór (VF – ventricular fibrillation) w okresach zaostrzenia choroby. W 75% przypadków pojawia się podczas codziennych czynności niezwiązanych z wysiłkiem, a jedynie w 3-5% w trakcie uprawiania sportu 44 . U starszych chorych częściej występują monomorficzne częstoskurcze komorowe (VT – ventricular tachycardia) 45 , wyzwalane w mechanizmie fali re-entry wokół obszarów uszkodzonego miokardium w fazie przewlekłej 4 .
Badania obrazowe
Najbardziej dostępnym badaniem obrazowym serca jest dwuwymiarowa echokardiografia przezklatkowa (TTE – transthoracic echocardiography). Przeprowadza się ją w poszukiwaniu odcinkowych lub globalnych zaburzeń kurczliwości prawej bądź lewej komory 6, 7 .
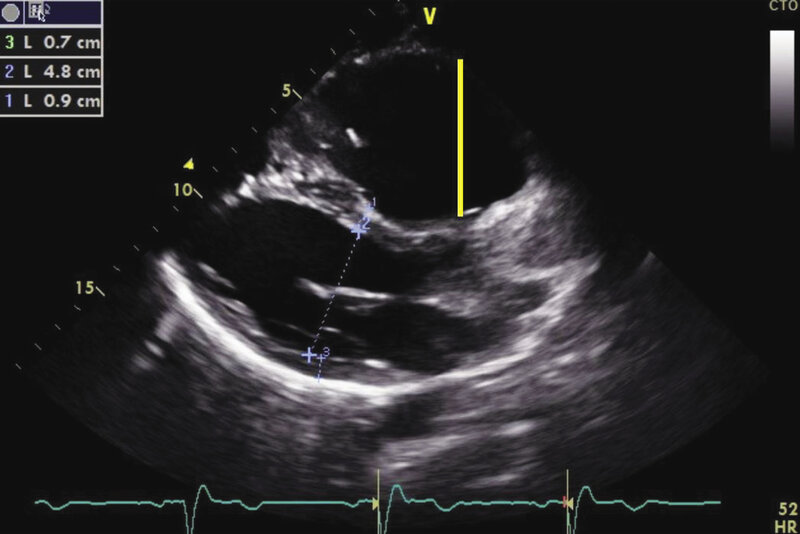
Rycina 5. Obraz echokardiograficzny arytmogennej kardiomiopatii prawokomorowej. Projekcja przymostkowa w osi długiej, zaznaczone poszerzenie wymiaru drogi odpływu prawej komory (ciągła żółta linia)
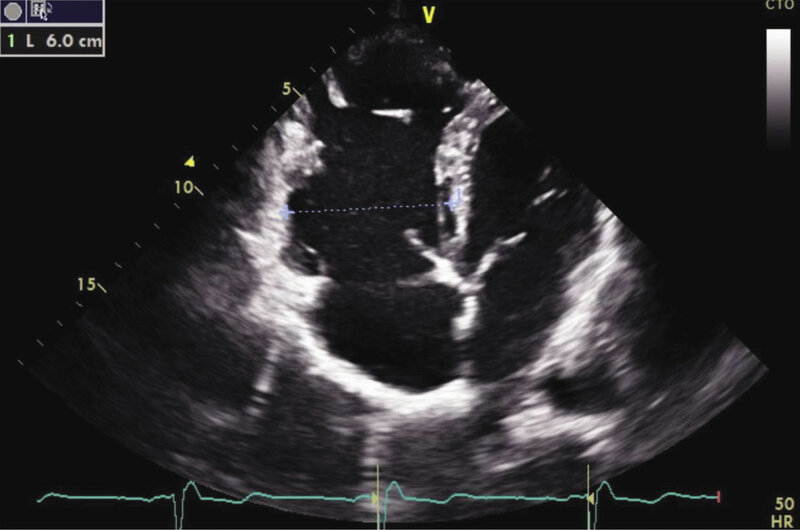
Rycina 6. Obraz echokardiograficzny arytmogennej kardiomiopatii prawokomorowej. Projekcja koniuszkowa czterojamowa, zaznaczone poszerzenie wymiaru drogi napływu prawej komory (niebieska przerywana linia)
Obrazowanie mięśnia sercowego za pomocą rezonansu magnetycznego, uzupełnione badaniem z późnym wzmocnieniem gadolinowym (LGE – late gadolinium enhancement), jest preferowanym badaniem obrazowym u pacjentów z podejrzeniem ARVC.
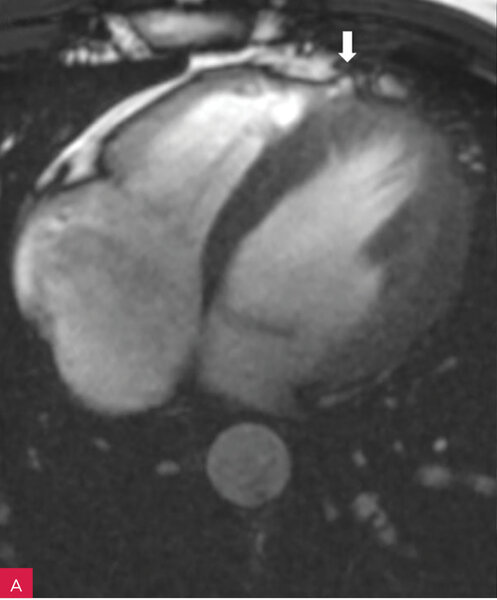
Rycina 7A, B. Rezonans magnetyczny serca u pacjenta z arytmogenną kardiomiopatią prawokomorową. Dyskinetyczny tętniak segmentu koniuszkowego wolnej ściany prawej komory (A). Progresja wielkości tętniaka w kontrolnym badaniu po upływie 12 miesięcy (B)
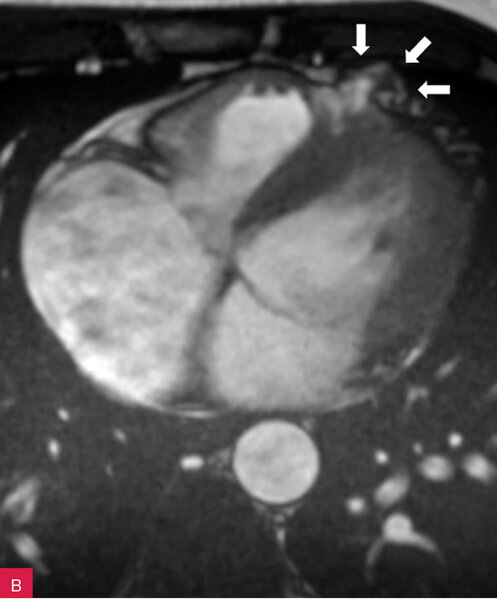
Rycina 7A, B. Rezonans magnetyczny serca u pacjenta z arytmogenną kardiomiopatią prawokomorową. Dyskinetyczny tętniak segmentu koniuszkowego wolnej ściany prawej komory (A). Progresja wielkości tętniaka w kontrolnym badaniu po upływie 12 miesięcy (B)
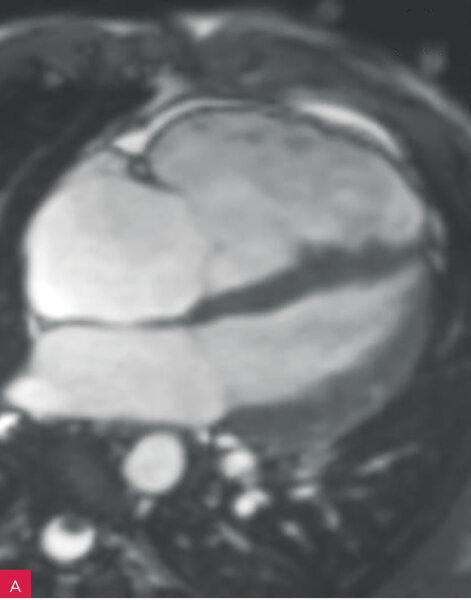
Rycina 8A-D. Rezonans magnetyczny serca u pacjenta z arytmogenną kardiomiopatią z zajęciem obu komór. Prawa komora powiększona z obniżoną funkcją skurczową i regionalnymi obszarami dyskinetycznymi – obrazy cine w fazie końcowoskurczowej (A, B). Tkanka włóknista w obrębie wolnej ściany prawej i lewej komory – obrazy opóźnionego wzmocnienia (C, D)
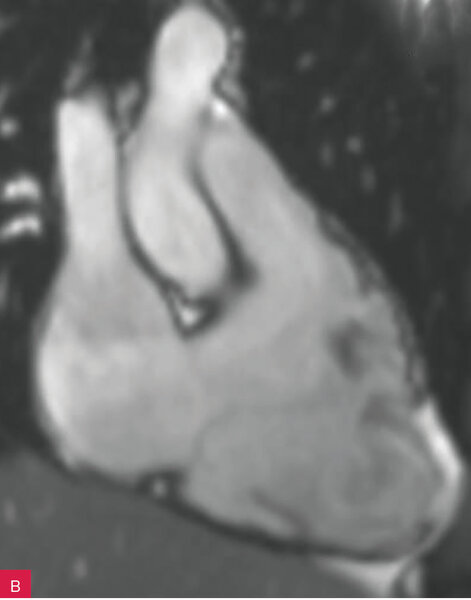
Rycina 8A-D. Rezonans magnetyczny serca u pacjenta z arytmogenną kardiomiopatią z zajęciem obu komór. Prawa komora powiększona z obniżoną funkcją skurczową i regionalnymi obszarami dyskinetycznymi – obrazy cine w fazie końcowoskurczowej (A, B). Tkanka włóknista w obrębie wolnej ściany prawej i lewej komory – obrazy opóźnionego wzmocnienia (C, D)
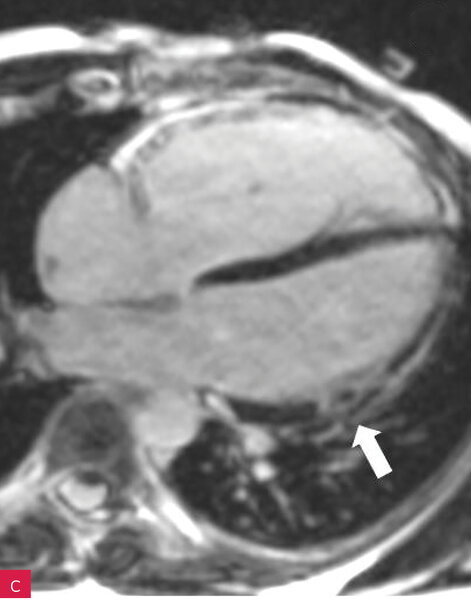
Rycina 8A-D. Rezonans magnetyczny serca u pacjenta z arytmogenną kardiomiopatią z zajęciem obu komór. Prawa komora powiększona z obniżoną funkcją skurczową i regionalnymi obszarami dyskinetycznymi – obrazy cine w fazie końcowoskurczowej (A, B). Tkanka włóknista w obrębie wolnej ściany prawej i lewej komory – obrazy opóźnionego wzmocnienia (C, D)
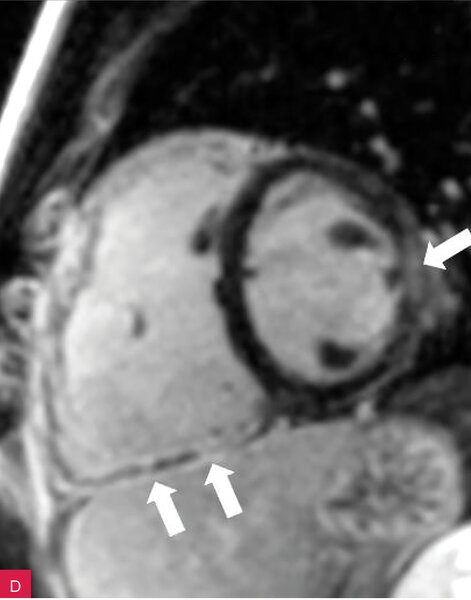
Rycina 8A-D. Rezonans magnetyczny serca u pacjenta z arytmogenną kardiomiopatią z zajęciem obu komór. Prawa komora powiększona z obniżoną funkcją skurczową i regionalnymi obszarami dyskinetycznymi – obrazy cine w fazie końcowoskurczowej (A, B). Tkanka włóknista w obrębie wolnej ściany prawej i lewej komory – obrazy opóźnionego wzmocnienia (C, D)
Wariantem diagnozowania obrazowego jest mapowanie elektroanatomiczne. Wydaje się narzędziem precyzyjnym, pozwalającym ustalić endokardialne obszary uszkodzenia elektrycznego. Z uwagi na inwazyjność eksperci nie zalecają jednak jego rutynowego stosowania. Wyjątkiem jest rozszerzenie wykonywanego badania elektrofizjologicznego lub ablacji przezskórnej 7 .
Badania genetyczne
Badanie genetyczne powinno się przeprowadzać u chorych z rozpoznaną ARVC (klasa zaleceń IB) 8 . Określenie patogennej mutacji może nie tylko uprawdopodobnić rozpoznanie lub uzupełnić ocenę ryzyka wystąpienia groźnych arytmii komorowych, lecz także ukierunkować prowadzenie badań przesiewowych wśród krewnych pacjenta 50 . Należy jednak pamiętać, że mutacje odpowiadające za ARVC mogą występować nawet u co piątej osoby ze zdrowym strukturalnie sercem 51 . Ponieważ badania nad alternatywnymi formami genowymi wciąż trwają, wskazanie mutacji odpowiedzialnej za chorobę może okazać się niemożliwe, a ujemny wynik genotypowania nie wyklucza rozpoznania.
Biopsja endomiokardialna
Biopsja endomiokardialna nie jest podstawowym narzędziem diagnostycznym w ARVC. Badanie to w większości przypadków zastąpiły nieinwazyjne metody obrazowania, które pozwalają dokładnie ocenić strukturę tkankową, bez narażania chorego na powikłania związane z inwazyjnymi działaniami. Nierównomierne zajęcie miokardium przez proces chorobowy, najczęściej jedynie na etapie zaawansowanym schorzenia, w okolicy przegrody międzykomorowej, stanowi o niskiej przydatności tej metody diagnostycznej. Jej czułość można poprawić, pobierając materiał z miejsca wybranego na podstawie mapy elektroanatomicznej 50 .
Ewolucja zaleceń diagnostycznych
Ze względu na brak patognomonicznego dla ARVC objawu postawienie diagnozy opiera się na skalach punktowych oceniających całość obrazu klinicznego. Ich pierwszą wersję, skupiającą się przede wszystkim na morfologii prawego serca, opublikowano w 1994 r. Uwzględniono w nich wywiad rodzinny, nieprawidłowości zapisu EKG, obrazowanie oraz wynik badania patomorfologicznego – biopsji endomiokardialnej, będącej ówczesnym złotym standardem diagnostycznym 37 . Postęp w zakresie metod diagnostycznych oraz poznanie wcześniejszych stadiów choroby doprowadziły do aktualizacji zaleceń w 2010 r. W celu zwiększenia czułości diagnostycznej zalecano dokładniejszą diagnostykę obrazową, elektrofizjologiczną oraz genetyczną 5 . Kolejna wersja wytycznych pojawiła się w 2020 r. Wówczas zwrócono uwagę na możliwość zajęcia procesem chorobowym także lewej komory serca. Wyróżniono pojęcie arytmogennej kardiomiopatii dzielącej się pod względem fenotypu na: dominującą prawokomorową (ARVC), obukomorową oraz dominującą lewokomorową (ALVC). Stało się to możliwe dzięki powszechnemu zastosowaniu rezonansu magnetycznego serca w diagnostyce chorych z ARVC, co pozwoliło na precyzyjną ocenę morfologiczną tkanek mięśnia sercowego bez potrzeby wykonywania biopsji endomiokardialnej 6 . Kryteria diagnostyczne zawarte w zaleceniach z 2010 r., a także ich aktualizację z 2020 r. cytowano w najnowszych wytycznych Europejskiego Towarzystwa Kardiologicznego (ESC – European Society of Cardiology) z 2023 r., koncentrujących się na diagnostyce i leczeniu kardiomiopatii 8 . Na początku 2024 r., opierając się na wynikach kolejnych doniesień literaturowych, European Task Force opublikowało stanowisko ekspertów będące rozwinięciem kryteriów diagnostycznych z Padwy („Padua criteria”) z 2020 r. 7 Na tej podstawie można postawić „pewną” diagnozę ARVC, gdy są spełnione: 2 kryteria większe lub 1 większe i 2 mniejsze, lub 4 mniejsze. Każde ze stwierdzanych kryteriów musi dotyczyć innej kategorii. Jeżeli postawienie pewnej diagnozy nie jest możliwe (non-define), chory powinien pozostać w regularnej obserwacji. Wynika to z faktu, że wraz z postępem schorzenia kolejne kryterium może zostać spełnione 5, 6, 7 . Fenotyp kardiomiopatii – dominująco prawokomorowy, lewokomorowy lub obukomorowy – określa się na podstawie kryteriów morfologiczno-funkcjonalnych i strukturalnych. Najnowsze kryteria diagnostyczne przedstawiono w tabeli 2.
Warto zauważyć, że spośród dostępnych skal diagnostycznych jedynie ta z 2010 r. została w pełni przebadana pod kątem czułości i swoistości. Co ciekawe, uniknięcie analizy późnych potencjałów oraz wywiadu rodzinnego zwiększyło szansę na postawienie trafnego rozpoznania do 97%, przy braku wpływu na czułość metody 32 . Zaproponowano także schemat diagnostyczny, w którym na etapie screeningu zastosowano 100% czułe i nieswoiste kryteria dotyczące zmian w zapisie EKG oraz monitorowania metodą holterowską. W kolejnym etapie rekomendowano przeprowadzanie badań bardziej swoistych, obejmujących echokardiografię przezklatkową, rezonans magnetyczny, badania genetyczne 32 .
Podsumowanie
ARVC to stosunkowo niedawno odkryta, rzadko występująca jednostka chorobowa, będąca przedmiotem zainteresowania wielu badaczy. Obraz kliniczny jest dynamiczny, co utrudnia, mimo prowadzonych badań, dokładne poznanie patofizjologii schorzenia. Niestety ARVC wiąże się ze zwiększonym ryzykiem występowania SCD. To obliguje lekarzy do zachowania czujności diagnostycznej oraz aktywnego poszukiwania chorych, u których ryzyko groźnych komorowych zaburzeń rytmu jest istotnie wyższe.
Abstract
Arrhythmogenic right ventricular cardiomyopathy: is it still difficult to diagnose?
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is associated with functional or structural abnormalities of the myocardium. Despite the growing understanding of the pathophysiology of this disease, there are still numerous uncertainties regarding not only the choice of optimal therapy, but also the establishment of a definitive diagnosis of ARVC. This paper presents the current state of knowledge on the diagnostic options based on the latest recommendations of cardiovascular societies and expert groups.
- 1. Elliott P, Andersson B, Arbustini E, et al. Classification of the cardiomyopathies: A position statement from the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2008;29(2):270-6
- 2. Marcus FI, Fontaine GH, Guiraudon G, et al. Right ventricular dysplasia: A report of 24 adult cases. Circulation 1982;65(2):384-98
- 3. Marcus FI. Guy Fontaine: a pioneer in electrophysiology. Clin Cardiol 1998;21(2):145-6
- 4. Corrado D, Basso C, Judge DP. Arrhythmogenic cardiomyopathy. Circ Res 2017;121(7):785-802
- 5. Marcus FI, McKenna WJ, Sherrill D, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: Proposed modification of the Task Force Criteria. Circulation 2010;121(13):1533-41
- 6. Corrado D, Perazzolo Marra M, et al. Diagnosis of arrhythmogenic cardiomyopathy: The Padua criteria. Int J Cardiol [Internet] 2020;319:106-14
- 7. Corrado D, Anastasakis A, Basso C, et al. Proposed diagnostic criteria for arrhythmogenic cardiomyopathy: European Task Force consensus report. Int J Cardiol 2024;395:131447
- 8. Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies: Developed by the task force on the management of cardiomyopathies of the European Society of Cardiology (ESC). Eur Heart J 2023;44(37):3503-626
- 9. Te Riele ASJM, James CA, Groeneweg JA, et al. Approach to family screening in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Eur Heart J 2016;37(9):755-63
- 10. James CA, Bhonsale A, Tichnell C, et al. Exercise increases age-related penetrance and arrhythmic risk in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. J Am Coll Cardiol 2013;62(14):1290-7
- 11. Corrado D, Basso C, Thiene G. Sudden cardiac death in young people with apparently normal heart. Cardiovasc Res 2001;50(2):399-408
- 12. Hulot JS, Jouven X, Empana JP, et al. Natural history and risk stratification of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation 2004;110(14):1879-84
- 13. Dalal D, Nasir K, Bomma C, et al. Arrythmogenic right ventricular cardiomiopathy. Circulation 2005;112:3823-32
- 14. Nava A, Bauce B, Basso C, et al. Clinical profile and long-term follow-up of 37 families with arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol 2000;36(7):2226-33
- 15. Lemola K, Brunckhorst C, Helfenstein U, et al. Predictors of adverse outcome in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy: Long term experience of a tertiary care centre. Heart 2005;91(9):1167-72
- 16. Krahn AD, Wilde AAM, Calkins H, et al. Arrhythmogenic Right Ventricular Cardiomyopathy. JACC Clin Electrophysiol 2022;8(4):533-53
- 17. Corrado D, Link MS, Calkins H. Arrhythmogenic Right Ventricular Cardiomyopathy. N Engl J Med 2017;376:61-72
- 18. James CA, Jongbloed JDH, Hershberger RE, et al. International Evidence Based Reappraisal of Genes Associated With Arrhythmogenic Right Ventricular Cardiomyopathy Using the Clinical Genome Resource Framework. Circ Genomic Precis Med 2021;14(3):E003273
- 19. Hoorntje ET, Te Rijdt WP, James CA, et al. Arrhythmogenic cardiomyopathy: Pathology, genetics, and concepts in pathogenesis. Cardiovasc Res 2017;113(12):1521-31
- 20. Groeneweg JA, Bhonsale A, James CA, et al. Clinical Presentation, Long-Term Follow-Up, and Outcomes of 1001 Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy Patients and Family Members. Circ Cardiovasc Genet 2015;8(3):437-46
- 21. Te Riele ASJM, James CA, Bhonsale A, et al. Malignant Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy with a normal 12-lead electrocardiogram: A rare but underrecognized clinical entity. Heart Rhythm [Internet] 2013;10(10):1484-91
- 22. Piccini JP, Nasir K, Bomma C, et al. Electrocardiographic findings over time in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Am J Cardiol 2005;96(1):122-6
- 23. Quarta G, Ward D, Tomé Esteban MT, et al. Dynamic electrocardiographic changes in patients with arrhythmogenic right ventricular cardiomyopathy. Heart 2010;96(7):516-22
- 24. Abou Jaoude S, Leclercq JF, Coumel P. Progressive ECG changes in arrhythmogenic right ventricular disease. Evidence for an evolving disease. Eur Heart J 1996;17(11):1717-22
- 25. Saguner AM, Ganahl S, Baldinger SH, et al. Usefulness of electrocardiographic parameters for risk prediction in arrhythmogenic right ventricular dysplasia. Am J Cardiol [Internet] 2014;113(10):1728-34
- 26. Peters S, Trümmel M, Koehler B. QRS fragmentation in standard ECG as a diagnostic marker of arrhythmogenic right ventricular dysplasia-cardiomyopathy. Heart Rhythm 2008;5(10):1417-21
- 27. Steriotis AK, Bauce B, Daliento L, et al. Electrocardiographic Pattern in Arrhythmogenic Right Ventricular Cardiomyopathy. Am J Cardiol [Internet] 2009;103(9):1302-8
- 28. Cox MGPJ, Nelen MR, Wilde AAM, et al. Activation delay and VT parameters in arrhythmogenic right ventricular dysplasia/cardiomyopathy: Toward improvement of diagnostic ECG criteria. J Cardiovasc Electrophysiol 2008;19(8):775-81
- 29. Nasir K, Bomma C, Tandri H, et al. Electrocardiographic features of arrhythmogenic right ventricular dysplasia/cardiomyopathy according to disease severity: A need to broaden diagnostic criteria. Circulation 2004;110(12):1527-34
- 30. Bhonsale A, James CA, Tichnell C, et al. Risk stratification in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. Circ Arrhythmia Electrophysiol 2013;6(3):569-78
- 31. De Lazzari M, Zorzi A, Cipriani A, et al. Relationship between electrocardiographic findings and cardiac magnetic resonance phenotypes in arrhythmogenic cardiomyopathy. J Am Heart Assoc 2018;7(22):1-11
- 32. Bosman LP, Cadrin-Tourigny J, Bourfiss M, et al. Diagnosing arrhythmogenic right ventricular cardiomyopathy by 2010 task force criteria: Clinical performance and simplified practical implementation. Europace 2020;22(5):787-96
- 33. Jain R, Dalal D, Daly A, et al. Electrocardiographic features of arrhythmogenic right ventricular dysplasia. Circulation 2009;120(6):477-87
- 34. Canpolat U, Kabakçi G, Aytemir K, et al. Fragmented QRS complex predicts the arrhythmic events in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. J Cardiovasc Electrophysiol 2013;24(11):1260-6
- 35. Pérez-Riera AR, Barbosa-Barros R, Daminello-Raimundo R, et al. Epsilon wave: A review of historical aspects. Indian Pacing Electrophysiol J 2019;19(2):63-7
- 36. Platonov PG, Calkins H, Hauer RN, et al. High interobserver variability in the assessment of epsilon waves: Implications for diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia. Heart Rhythm [Internet] 2016;13(1):208-16
- 37. McKenna WJ, Thiene G, Nava A, et al. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomiopathy. Br Heart J 1994;71(3):215-8
- 38. Peters S, Trümmel M. Diagnosis of arrhythmogenic right ventricular dysplasia-cardiomyopathy: Value of standard ECG revisited. Ann Noninvasive Electrocardiol 2003;8(3):238-45
- 39. Camm CF, James CA, Tichnell C, et al. Prevalence of atrial arrhythmias in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Heart Rhythm [Internet] 2013;10(11):1661-8
- 40. Zghaib T, Bourfiss M, van der Heijden JF, et al. Atrial Dysfunction in Arrhythmogenic Right Ventricular Cardiomyopathy. Circ Cardiovasc Imaging 2018;11(9):e007344
- 41. Akdis D, Saguner AM, Shah K, et al. Sex hormones affect outcome in arrhythmogenic right ventricular cardiomyopathy/dysplasia: Froma stemcell derived cardiomyocyte-based model to clinical biomarkers of disease outcome. Eur Heart J 2017;38(19):1498-508
- 42. D’Silva A, Papadakis M. Sudden cardiac death in athletes. Eur Cardiol Rev 2015;10(1):48-53
- 43. Corrado D, Basso C, Schiavon M, et al. Screening for Hypertrophic Cardiomyopathy in Young Athletes. New Engl J Med 1998;339(6):364-9
- 44. Tabib A, Loire R, Chalabreysse L, et al. Circumstances of Death and Gross and Microscopic Observations in a Series of 200 Cases of Sudden Death Associated with Arrhythmogenic Right Ventricular Cardiomyopathy and/or Dysplasia. Circulation 2003;108(24):3000-5
- 45. Bhonsale A, Groeneweg JA, James CA, et al. Impact of genotype on clinical course in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated mutation carriers. Eur Heart J 2015;36(14):847-55
- 46. Malik N, Win S, James CA, et al. Right ventricular strain predicts structural disease progression in patients with arrhythmogenic right ventricular cardiomyopathy. J Am Heart Assoc 2020;9(7):1-9
- 47. Addetia K, Mazzanti A, Maragna R, et al. Value of 3D echocardiography in the diagnosis of arrhythmogenic right ventricular cardiomyopathy. Eur Heart J – Cardiovasc Imaging [Internet] 2023;24(5):664-77
- 48. Marcus F, Basso C, Gear K, et al. Pitfalls in the Diagnosis of Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia. Am J Cardiol 2010;105(7):1036-9
- 49. Rastegar N, Burt JR, Corona-Villalobos CP, et al. Cardiac MR findings and potential diagnostic pitfalls in patients evaluated for arrhythmogenic right ventricular cardiomyopathy. Radiographics 2014;34(6):1553-71
- 50. Towbin JA, McKenna WJ, Abrams DJ, et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm [Internet] 2019;16(11):e301-72
- 51. Kapplinger JD, Landstrom AP, Salisbury BA, et al. Distinguishing arrhythmogenic right ventricular cardiomyopathy/dysplasia-associated mutations from background genetic noise. J Am Coll Cardiol 2011;57(23):2317-27




 Dodaj do ulubionych
Dodaj do ulubionych
