



Co znajdziesz w artykule?
- Omówienie wybranych typów kardiomiopatii i ich charakterystyka
- Wybrane badania diagnostyczne stosowane w procesie diagnostycznym kardiomiopatii
- Rola biopsji mięśnia sercowego w procesie diagnostycznym kardiomiopatii
Spis treści
Kryteria rozpoznania oraz klasyfikacja kardiomiopatii wielokrotnie się zmieniały w ciągu ostatnich lat. Najnowsza definicja kardiomiopatii została przedstawiona w 2023 roku przez European Society of Cardiology. Według niej kardiomiopatia to choroba mięśnia sercowego, która charakteryzuje się jego nieprawidłową strukturą i funkcją, bez obecności choroby niedokrwiennej serca, nadciśnienia tętniczego, choroby zastawkowej serca oraz wrodzonej wady serca, o nasileniu wystarczającym do wywołania
obserwowanej dysfunkcji 1 . Najczęściej stosowany podział bazujący na fenotypie wyróżnia 5 typów kardiomiopatii (tab. 1):
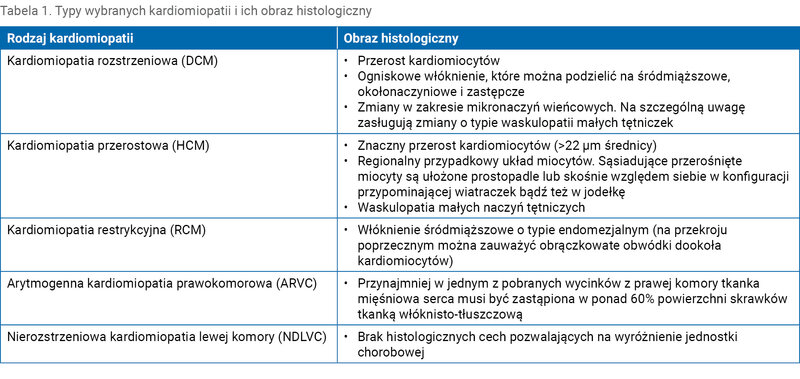
Tabela 1. Typy wybranych kardiomiopatii i ich obraz histologiczny
- kardiomiopatię rozstrzeniową (DCM – dilated cardiomyopathy)
- kardiomiopatię przerostową (HCM – hypertrophic cardiomyopathy)
- kardiomiopatię restrykcyjną (RCM – restrictive cardiomyopathy)
- arytmogenną kardiomiopatię prawokomorową (ARVC – arrhythmogenic right ventricular cardiomyopathy)
- nierozstrzeniową kardiomiopatię lewej komory (NDLVC – non-dilated left ventricular cardiomyopathy).
Podział kardiomiopatii na podstawie ich fenotypu uwzględnia cechy morfologiczne, a także wskaźniki kliniczne serca. Kardiomiopatie można również podzielić na rodzinne (uwarunkowane genetycznie) oraz niewystępujące w rodzinie (nieuwarunkowane genetycznie). W ustaleniu rozpoznania typu kardiomiopatii nadrzędne pozostają parametry kliniczne, a także precyzyjnie zebrany wywiad, w tym również wywiad rodzinny. Ocena morfologiczna dodatkowo pozwala na określenie stopnia zmian patologicznych w mięśniu sercowym oraz ich zaawansowania.
Biopsja mięśnia sercowego
Biopsja mięśnia sercowego, zwana biopsją endomiokardialną, została wprowadzona do badań przyżyciowych w kardiologii w roku 1963 i do chwili obecnej stanowi cenną metodę wspomagającą postępowanie diagnostyczno-lecznicze u chorych ze schorzeniami mięśnia sercowego. Niemniej ze względu na to, że jest to badanie inwazyjne obarczone ryzykiem powikłań zabiegowych, wykorzystuje się je wybiórczo do pogłębienia diagnostyki kardiomiopatii. Należy jednak zaznaczyć, że wraz z rozwojem technologicznym oraz doświadczeniem zespołów wykonujących tę procedurę ryzyko powikłań zostało zredukowane do minimum i nie przekracza 1% 2 . W wybranych przypadkach biopsja endomiokardialna stanowi metodę pozwalającą na ustalenie ostatecznego rozpoznania. Biopsja mięśnia sercowego umożliwia wykluczenie bądź potwierdzenie komponenty zapalnej, obecności zmian spichrzeniowych, na przykład amyloidozy bądź sarkoidozy, jak również określenie stopnia przebudowy mięśnia sercowego. Badanie wycinków mięśnia sercowego oprócz wyżej wymienionych wskaźników patologii coraz częściej uwzględnia też ocenę stopnia zmian waskulopatycznych, stresu oksydacyjnego oraz zaburzeń hemostazy tkankowej w wycinkach mięśnia sercowego. Wszystkie powyżej wymienione wskaźniki odgrywają istotną rolę w określeniu potencjalnych czynników etiopatogenetycznych schorzenia, a także pozwalają na dobór optymalnego postępowania leczniczego, w tym na podjęcie decyzji o wykonaniu przeszczepienia serca 3 .
Rodzaje kardiomiopatii oraz ich histologiczna prezentacja
Kardiomiopatia rozstrzeniowa
Najczęstszą, bo występującą w prawie 60% przypadków, kardiomiopatią jest DCM. Ta forma kardiomiopatii może zarówno mieć podłoże genetyczne, jak i być skutkiem oddziaływania czynników infekcyjnych, toksycznych bądź środowiskowych. Występowanie DCM o etiologii genetycznej jest uwarunkowane wieloma mutacjami genów białek sarkomeru (geny MYH7, TNNC1, TTN), otoczki jądrowej (gen LMNA), a także białek cytoszkieletu (gen DES, FNLC). Forma nabyta DCM jest z kolei najczęściej powiązana z czynnikami infekcyjnymi. Należą do nich przede wszystkim wirusy (Coxsackie, adenowirusy, wirusy grypy). Wśród czynników toksycznych wymienia się alkohol i alkaloidy (kokaina) oraz przyjmowanie niektórych leków, takich jak leki cytostatyczne (antracykliny) 4, 5 .
Dominującą formą klinicznej prezentacji DCM jest skurczowa niewydolność serca objawiająca się pogorszeniem tolerancji wysiłku, dusznością, orthopnoe czy też obrzękami obwodowymi. W fazie zaostrzenia niewydolności serca pojawia się także zastój w krążeniu małym skutkujący obecnością zmian osłuchowych (trzeszczeniami) nad polami płucnymi. Zmiany osłuchowe mogą wystąpić również podczas osłuchiwania pracy zastawek serca – u chorego może być słyszalna niedomykalność zastawki mitralnej, jak również zastawki trójdzielnej.
Podstawową rolę w ocenie struktury i funkcji serca w przebiegu DCM odgrywają badanie echokardiograficzne (UKG) oraz rezonans magnetyczny serca (MR – magnetic resonance). W badaniu echokardiograficznym kardiomiopatia rozstrzeniowa manifestuje się jedno- lub obukomorową progresywną rozstrzenią wraz ze zmniejszeniem grubości ściany serca (ryc. 1) oraz towarzyszącym upośledzeniem czynności skurczowej serca prowadzącym do spadku frakcji wyrzutowej, upośledzeniem odkształcania mięśnia sercowego oraz pogorszeniem parametrów pracy mięśnia sercowego. Na skutek rozstrzeni lewej komory serca i towarzyszącego poszerzenia pierścienia mitralnego często pojawia się niedomykalność zastawki mitralnej, a czasem również zastawki trójdzielnej.
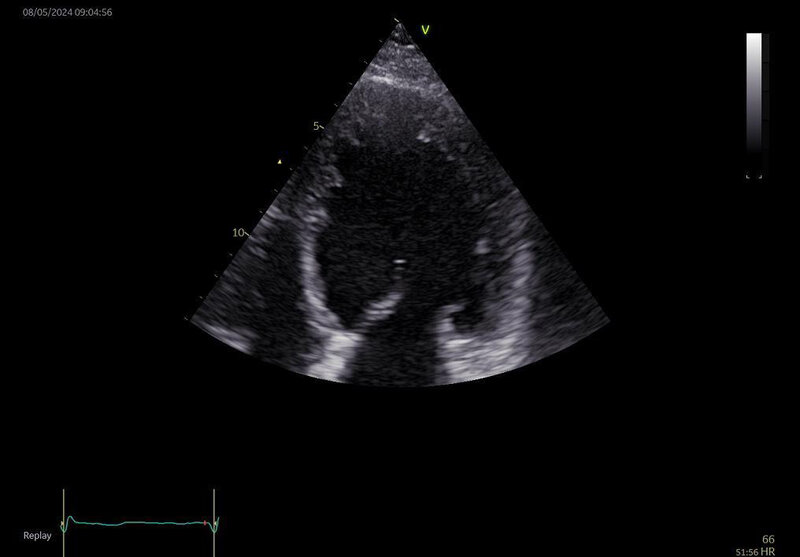
Rycina 1. Badanie echokardiograficzne. Kardiomiopatia rozstrzeniowa. Powiększona i zaokrąglona lewa komora
Dodatkowych informacji o etiologii i nasileniu zmian w mięśniu sercowym dostarcza rezonans magnetyczny serca. Jest to metoda dokładniejsza od echokardiografii w ocenie objętości skurczowej i rozkurczowej lewej komory serca, która pozwala na uzyskanie wiarygodniejszej oceny frakcji wyrzutowej lewej komory serca. Tym samym możliwe jest wykrycie znacznie wcześniej spadku frakcji wyrzutowej oraz śledzenie jej niewielkich wahań. Ponadto MR pozwala na charakterystykę samego mięśnia sercowego (ryc. 2). Do uwidocznienia różnic w charakterze tkanki mięśnia sercowego stosuje się zestaw rozmaitych sekwencji oraz obrazowanie metodą późnego wzmocnienia kontrastowego (LGE – late gadolinium enhancement), dzięki któremu można zidentyfikować obszary włóknienia oraz martwicy. W DCM są obserwowane liniowe lub ogniskowe wzmocnienia po podaniu środka kontrastowego, które występują przede wszystkim w warstwie śródmięśniowej w obszarach mięśnia sercowego nienależących do dorzecza określonej tętnicy wieńcowej.
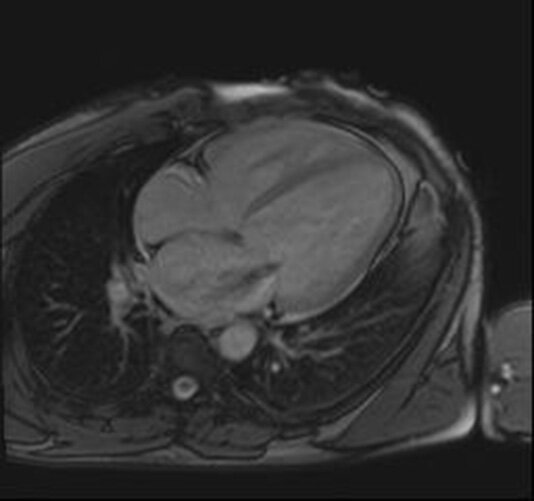
Rycina 2. Rezonans magnetyczny. Kardiomiopatia rozstrzeniowa. Rozstrzeń lewej komory z widocznym ścieńczeniem ściany mięśnia sercowego
Poprzez zastosowanie w badaniu MR gadolinowego środka kontrastowego z jego późnym wzmocnieniem uzyskuje się obrazowanie nieodwracalnego włóknienia zastępczego. Rozproszone włóknienie śródmiąższowe jest natomiast lepiej uwidaczniane za pomocą mapowania T1 6 . Technika mapowania T2 pozwala ponadto na wykazanie nadmiernej ilości płynu w tkankach mięśnia sercowego.
Biopsja mięśnia sercowego najczęściej jest wykonywana w celu potwierdzenia lub wykluczenia czynnika zapalnego (ryc. 3) w etiopatogenezie tego schorzenia 3 . Na podstawie uzyskanego wyniku wyróżnia się podtyp kardiomiopatii rozstrzeniowej, jaką jest kardiomiopatia zapalna (inflammatory dilated cardiomyopathy) 7 . Wykazanie obecności reakcji zapalnej pozwala na wdrożenie terapii przeciwzapalnej 8 . Zwykle zmiany w obrazie histopatologicznym obserwowane w wycinkach endomiokardialnych w przebiegu DCM są niespecyficzne. Dominują przerost kardiomiocytów oraz ogniskowe włóknienie, które można podzielić na śródmiąższowe, okołonaczyniowe i zastępcze 9 . Coraz częściej zwraca się także uwagę na zmiany w zakresie mikronaczyń wieńcowych. Szczególnie interesujące są zmiany o typie waskulopatii małych (końcowych) tętniczek. Ich obecność może wskazywać na podłoże nadciśnieniowe zmian patologicznych, a także potwierdzać istotną rolę zaburzeń struktury i funkcji mikrokrążenia w etiopatogenezie tego typu kardiomiopatii.
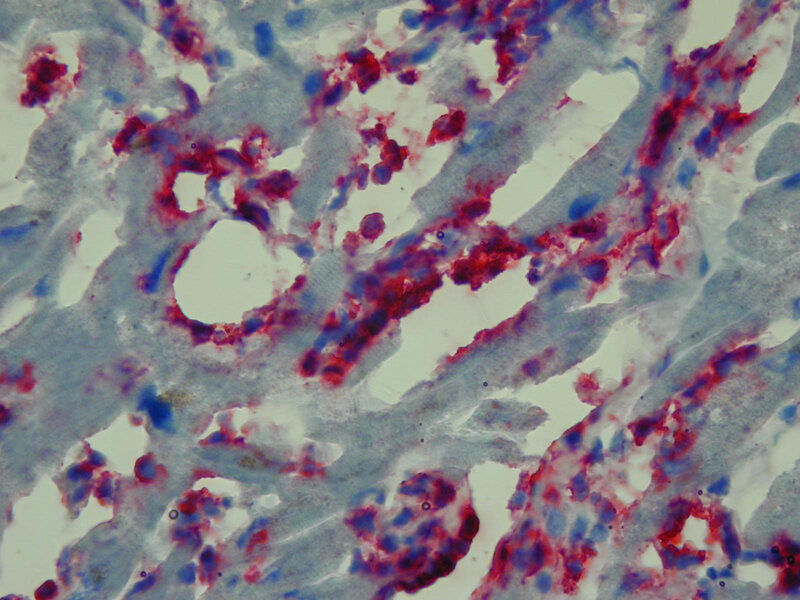
Rycina 3. Biopsja endomiokardialna. Kardiomiopatia rozstrzeniowa zapalna. Obecny nasilony naciek limfocytarny składający się z komórek CD3(+) (czerwony kolor). Reakcja immunohistochemiczna na skrawkach mrożeniowych
Kardiomiopatia przerostowa
Kardiomiopatia przerostowa charakteryzuje się najczęściej asymetrycznym przerostem mięśnia sercowego w zakresie przegrody międzykomorowej lub też segmentów koniuszkowych lewej komory serca, któremu towarzyszą zaburzenia funkcji rozkurczowej mięśnia sercowego. Najczęstszą jej przyczyną jest mutacja genu kodującego białko sarkomeru sercowego (np. β-miozyny, białka C wiążącego miozynę, troponiny T) 10 . Choroba objawia się dusznością wysiłkową, kołataniem serca, bólami w klatce piersiowej oraz omdleniami. U części chorych przebiega bezobjawowo i pierwszą jej manifestacją może być nagłe zatrzymanie krążenia 11 . W badaniu przedmiotowym u chorych ze zwężeniem drogi odpływu z lewej komory (LVOT – left ventricular outflow tract) można usłyszeć szmer skurczowy wzdłuż lewego brzegu mostka promieniujący do prawego górnego brzegu mostka i koniuszka serca oraz stwierdzić chybkie, dwubitne tętno.
W badaniu echokardiograficznym HCM charakteryzuje się asymetrycznym pogrubieniem ściany lewej komory (≥15 mm), którego nie można wyjaśnić jedynie jej nieprawidłowym obciążeniem (ryc. 4).
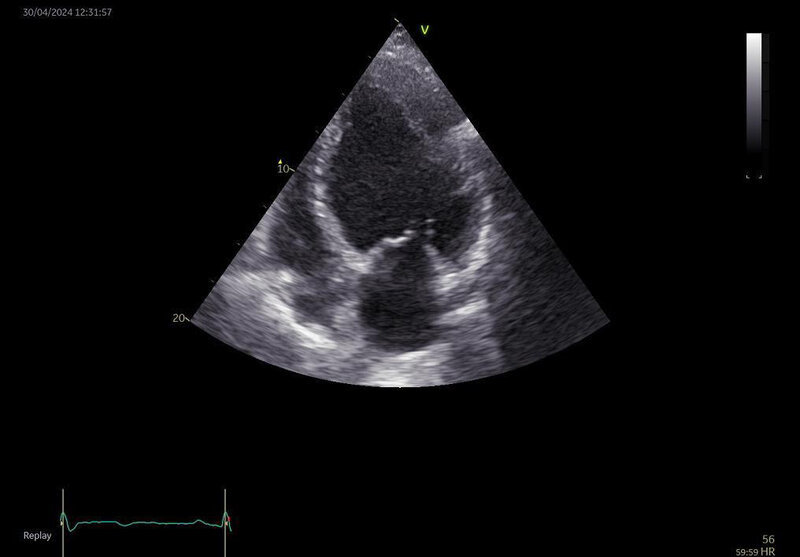
Rycina 4. Obraz echokardiograficzny. Kardiomiopatia przerostowa z przerostem segmentów koniuszkowych
U części chorych oprócz asymetrycznego przerostu lewej komory można również stwierdzić zwężenie LVOT oraz wynikającą z tego niedomykalność zastawki mitralnej. Zwężenie drogi odpływu lewej komory określa się jako szczytowy gradient między LVOT a aortą mierzony przy użyciu obrazowania dopplerowskiego, który wynosi ≥30 mmHg. Pacjent z LVOT ≥50 mmHg oraz występującymi omdleniami wysiłkowymi kwalifikuje się do leczenia inwazyjnego 10 .
U chorych zakwalifikowanych do leczenia inwazyjnego poza oceną w badaniu echokardiograficznym należy również wykonać MR serca. Badanie to odgrywa istotną rolę w diagnostyce HCM (ryc. 5) w zakresie oceny stopnia zwłóknienia serca z wykorzystaniem sekwencji LGE. Zwłókniałe obszary charakteryzują się ekspansją pozakomórkową, dlatego gromadzący się tam gadolin wykaże nadmierne wzmocnienie w porównaniu ze zdrowym mięśniem sercowym 13 . Należy jednak pamiętać, że obrazowanie późnego wzmocnienia po podaniu gadolinu wiąże się z większym ryzykiem wystąpienia arytmii komorowych u pacjenta z HCM 14 .
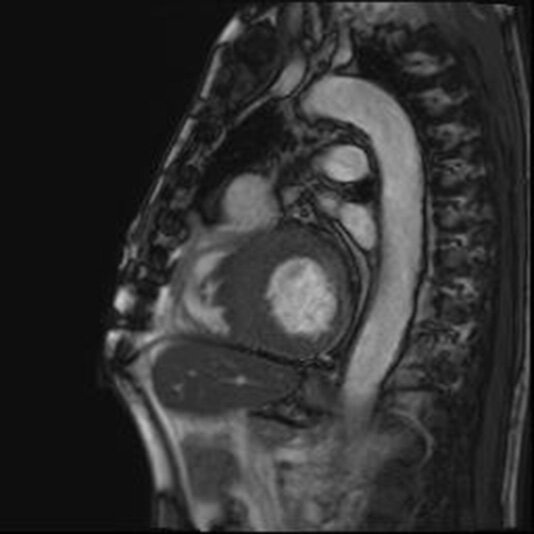
Rycina 5. Rezonans magnetyczny. Kardiomiopatia przerostowa. Cechy znacznego pogrubienia wymiarów ściany lewej komory
Biopsja endomiokardialna u pacjentów z HCM jest pomocna w kwalifikowaniu ich do przeszczepienia serca, jednak przede wszystkim pozwala na wykluczenie współistniejącej komponenty zapalnej będącej możliwą przyczyną progresji choroby w kierunku kardiomiopatii rozstrzeniowej. Obraz histologiczny w kardiomiopatii przerostowej charakteryzuje się znacznym przerostem kardiomiocytów (>22 μm średnicy), a także ich regionalnym przypadkowym układem 15 . Sąsiadujące przerośnięte miocyty są ułożone prostopadle lub skośnie względem siebie w konfiguracji przypominającej wiatraczek bądź też w jodełkę (ryc. 6) 15 . U około 70% chorych jedną z istotnych cech morfologicznych jest waskulopatia małych naczyń tętniczych.
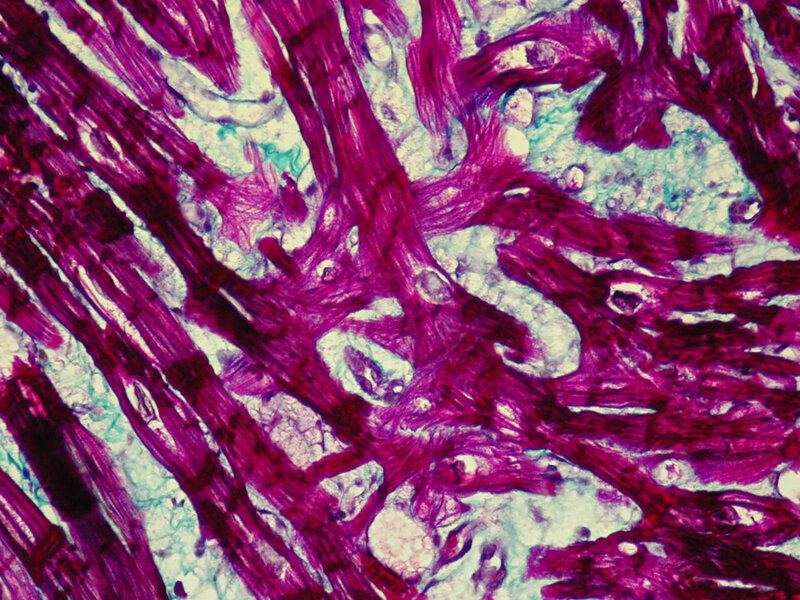
Rycina 6. Biopsja endomiokardialna. Kardiomiopatia przerostowa. Chaotyczny przebieg włókien mięśniowych (czerwony kolor) w reakcji histochemicznej metodą trójbarwną Massona (włókna kolagenowe zabarwione na kolor zielony)
Kardiomiopatia restrykcyjna
Kardiomiopatia restrykcyjna jest uważana za jedną z najrzadziej występujących chorób mięśnia sercowego oraz jedną z najtrudniejszych do zdiagnozowania 16 . Schorzenie to może mieć podłoże zarówno idiopatyczne, jak i wtórne, na przykład w wyniku amyloidozy, hemochromatozy bądź sarkoidozy serca (ryc. 7). Zarówno włóknienie, jak i odkładanie się w sercu substancji spichrzeniowych doprowadza stopniowo do wzrostu sztywności mięśnia sercowego skutkującej w końcowej fazie niewydolnością prawej (objętościowej) komory serca. W tej fazie choroby w badaniu przedmiotowym można stwierdzić obrzęki obwodowe, powiększenie wątroby, objaw wątrobowo-szyjny, wodobrzusze i związane z tym zaburzenia ze strony przewodu pokarmowego. Należy jednak podkreślić, że ten typ kardiomiopatii nie ma swoistych objawów. W diagnostyce RCM są wykorzystywane różne metody i techniki diagnostyczne.
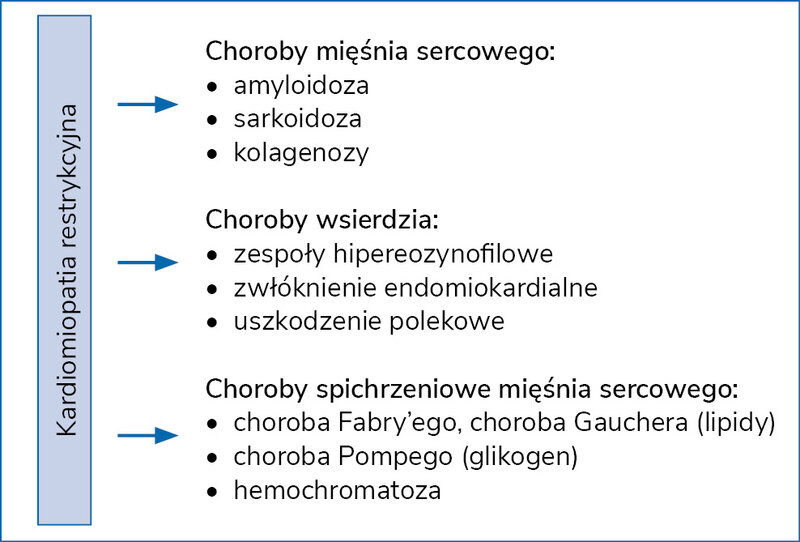
Rycina 7. Możliwe podłoże kardiomiopatii restrykcyjnej
Należą do nich: ocena echokardiograficzna, techniki rezonansu magnetycznego, tomografia emisyjna pojedynczego fotonu (SPECT – single photon emission computed tomography) całego ciała, a w niektórych przypadkach w celu oceny zmian restrykcyjnych pomocne jest cewnikowanie prawego serca z pomiarem ciśnień wewnątrzsercowych.
W badaniu echokardiograficznym RCM charakteryzuje się przede wszystkim zaburzoną funkcją rozkurczową serca wskazującą na podwyższone ciśnienie napełniania lewej komory serca. Pomimo często prawidłowej frakcji wyrzutowej lewej komory serca obrazowanie odkształcenia może wykazywać zaburzenia. Co więcej, w przypadku niektórych schorzeń prowadzących do RCM opisano charakterystyczne wzorce zaburzeń odkształcania, np. zachowane odkształcanie wyłącznie segmentów koniuszkowych lewej komory serca w amyloidozie (ryc. 8) lub też zaburzenia odkształcania w zakresie ściany tylnej w chorobie Fabry’ego. Inne częste objawy to poszerzenie przedsionków i przerost mięśnia sercowego. Chorzy z RCM zazwyczaj mają poszerzone jamy przedsionków z zachowanymi, nieposzerzonymi jamami jednej bądź obu komór serca. Ponadto u pacjentów może występować zwiększona bądź prawidłowa grubość ściany lewej komory 17 .
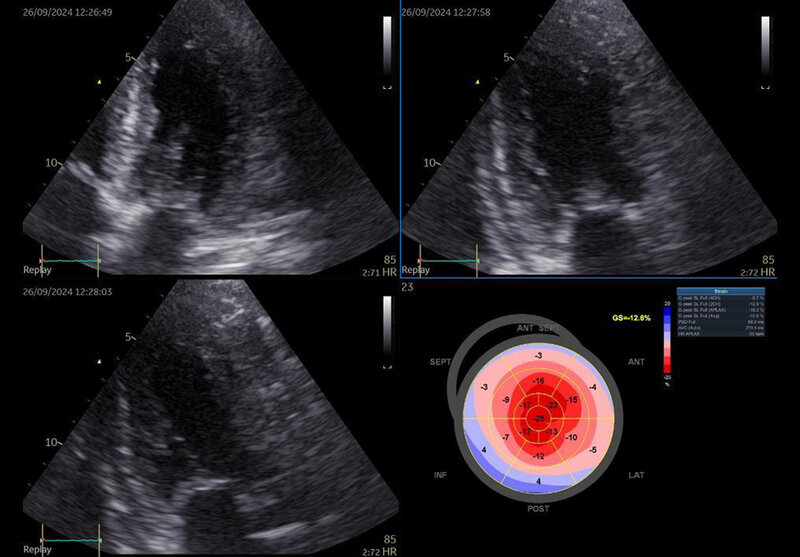
Rycina 8. Badanie echokardiograficzne. Kardiomiopatia restrykcyjna na podłożu amyloidozy – charakterystyczne zachowanie odkształcania w zakresie segmentów koniuszkowych (apical sparing)
W badaniu MR w zależności od podłoża zmian chorobowych obserwuje się różne wzorce obrazowania (ryc. 9). Przykładowo w przebiegu amyloidozy serca stwierdza się rozproszone podwsierdziowe późne wzmocnienie gadolinowe 17 . Badanie MR serca jest także przydatne w celu odróżnienia RCM oraz zaciskającego zapalenia osierdzia, których objawy kliniczne są podobne.
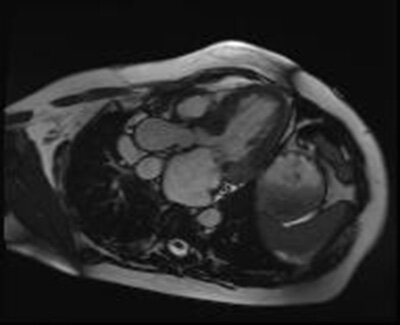
Rycina 9. Rezonans magnetyczny. Kardiomiopatia restrykcyjna na podłożu choroby Fabry’ego
U części chorych z RCM dużą rolę w ustaleniu potencjalnego czynnika etiologicznego odgrywa biopsja endomiokardialna 17 . Pozwala ona przede wszystkim na potwierdzenie lub wykluczenie zmian naciekowych oraz spichrzeniowych serca. Do pierwszej grupy należy gromadzenie się złogów amyloidu (amyloidoza serca) oraz żelaza (hemochromatoza). Drugi typ zmian obejmuje przede wszystkim choroby spichrzeniowe glikogenu i związków lipidowych 18 . W przypadku amyloidozy biopsja endomiokardialna pozwala nie tylko potwierdzić występowanie samej choroby, lecz także określić typ amyloidozy, co decyduje o włączeniu odpowiedniej terapii enzymatycznej (ryc. 10). Obraz histopatologiczny RCM charakteryzuje się swoistym włóknieniem śródmiąższowym. W przekroju poprzecznym można zauważyć obrączkowate obwódki dookoła kardiomiocytów 3 .
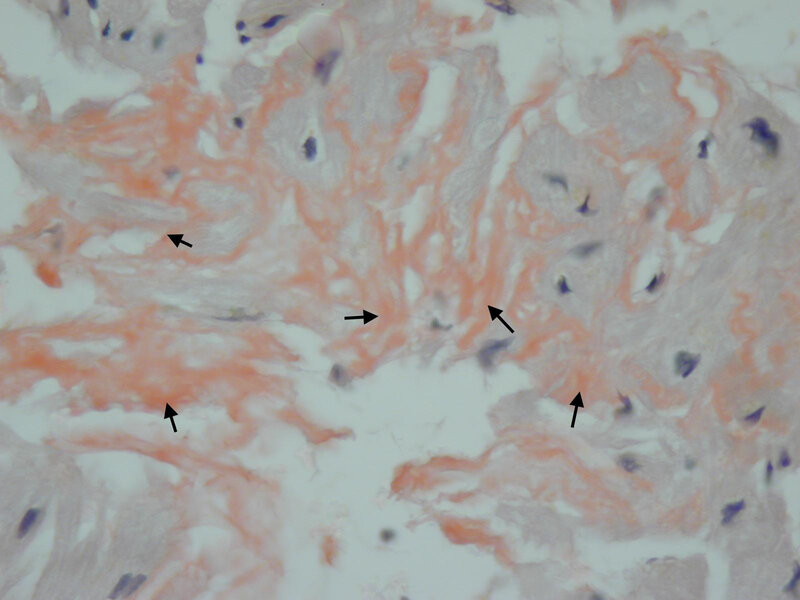
Rycina 10. Biopsja endomiokardialna. Kardiomiopatia restrykcyjna. Amyloidoza serca w barwieniu metodą czerwieni Kongo (strzałki – amyloid zabarwiony na kolor pomarańczowy)
Arytmogenna kardiomiopatia prawokomorowa
Arytmogenna kardiomiopatia prawokomorowa jest chorobą dziedziczoną autosomalnie dominująco, w której dochodzi do zastępowania włókien mięśniowych serca tkanką włóknisto-tłuszczową, co w efekcie powoduje arytmie komorowe. Zmiany zachodzące w mięśniu sercowym rozwijają się stopniowo. Pierwsze zmiany następują w warstwie nasierdziowej, a ostatecznie dotyczą wszystkich warstw mięśnia sercowego 19 . Choroba najczęściej występuje u młodych osób, a pierwszym objawem może być krótkotrwała utrata przytomności związana z wysiłkiem fizycznym 18 . Chorzy oprócz zagrażających życiu arytmii komorowych mają również objawy niewydolności serca prawokomorowej 19 . Poza takimi objawami, jak kołatanie serca czy zawroty głowy, może również dojść do nagłego zatrzymania krążenia w przebiegu zaburzeń rytmu pracy komór, które najczęściej występują u młodych sportowców 19 . Na podstawie International Task Force 2010, na które składa się zapis EKG, badanie echokardiograficzne, MR serca oraz biopsja endomiokardialna, można ustalić ostateczne rozpoznanie. W każdym z wcześniej wspomnianych badań ARVC ma charakterystyczne zmiany.
W badaniu echokardiograficznym dla ARVC typowe są nieregularne poszerzenie drogi odpływu prawej komory oraz ścieńczenie grubości ściany prawej komory (ryc. 11). Można również zauważyć obszary dyskinetyczne, które wynikają z zastąpienia tkanki mięśniowej przez tkankę włóknisto-tłuszczową. Istotną cechą jest także obecność pojedynczych lub mnogich tętniaków 19 .
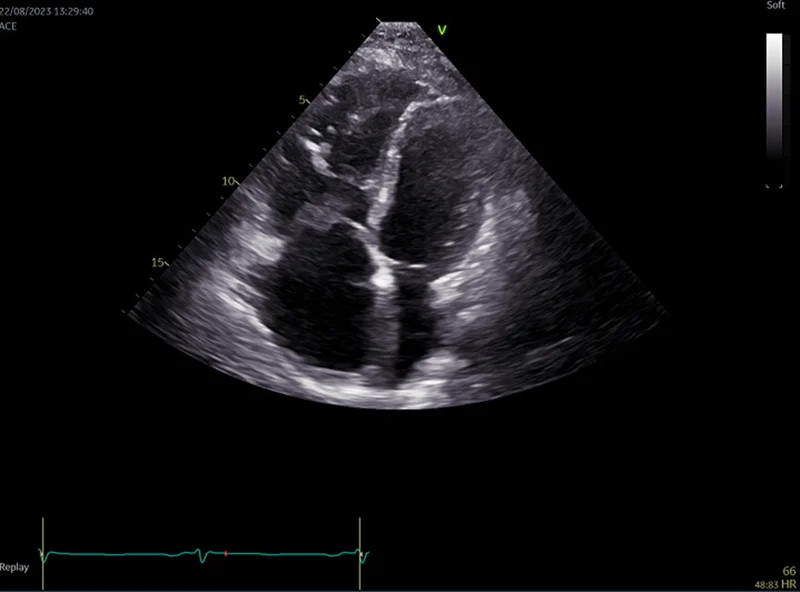
Rycina 11. Badanie echokardiograficzne. Arytmogenna kardiomiopatia prawej komory. Rozstrzeń prawej komory
W MR serca obserwuje się odcinkową akinezę lub dyskinezę prawej komory, a ponadto dyssynchronię skurczu oraz jeden z następujących parametrów:
- stosunek objętości końcoworozkurczowej prawej komory (RV – right ventricle) do powierzchni ciała pacjenta (BSA – body surface area) od ≥100 do <110 ml/m 2 (mężczyźni) lub od ≥90 do <100 ml/m 2 (kobiety) albo
- frakcja wyrzutowa RV od >40% do ≤45%.
W przypadku biopsji endomiokardialnej uważa się, że przynajmniej w jednym z pobranych wycinków z prawej komory tkanka mięśniowa serca musi być zastąpiona w >60% powierzchni skrawków tkanką włóknisto-tłuszczową (ryc. 12) 3, 18, 19 .
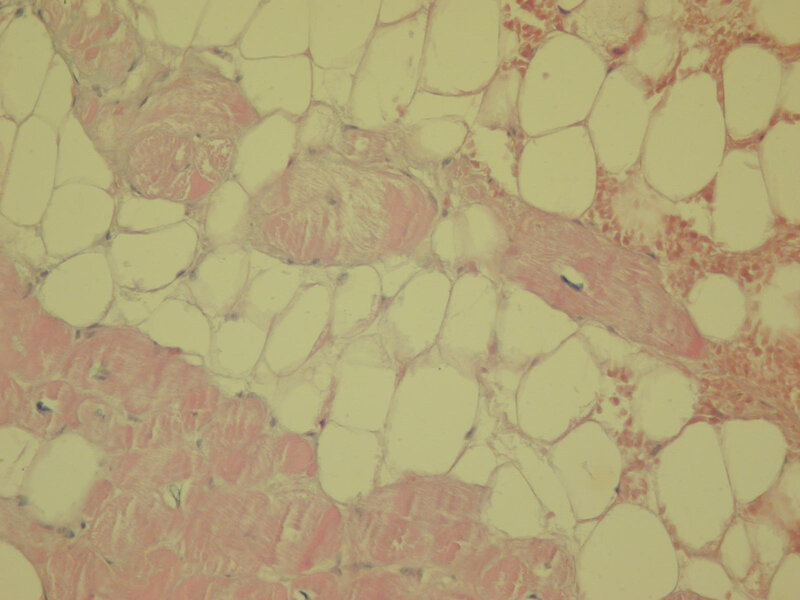
Rycina 12. Biopsja endomiokardialna. Arytmogenna kardiomiopatia prawej komory. Pomiędzy komórkami mięśniowymi obecna tkanka tłuszczowa. Barwienie histochemiczne (hematoksylina i eozyna)
Nierozstrzeniowa kardiomiopatia lewej komory
Nierozstrzeniowa kardiomiopatia lewej komory jest stosunkowo niedawno zaproponowaną grupą kardiomiopatii. W jej przebiegu chorzy mają zachowaną prawidłową funkcję skurczową. Jej cechą są zachowane wymiary jam serca z obecnością blizny o etiologii innej niż niedokrwienie czy też związaną z przebudową tłuszczową 20 . Sugeruje się, że nowo wyodrębniona grupa kliniczna powstała na potrzeby pacjentów, których przypadki zaklasyfikowano jako subkliniczna DCM lub potencjalnie rozwijająca się w kierunku DCM, niespełniających wszystkich diagnostycznych kryteriów 21 . Niektórzy nawet nazywają nierozstrzeniową kardiomiopatię lewej komory łagodniejszą wersją DCM, jednak ostatnie badania wykazały, że chorzy z NDLVC nie mogą być traktowani w kategorii łagodniejszej wersji choroby (ryc. 13) 21 . U chorych z NDLVC stwierdza się wyższe ryzyko wystąpienia migotania przedsionków, cukrzycy oraz hiperlipidemii 20 . Pomimo rosnącego zainteresowania tą jednostką chorobową nadal posiadamy bardzo mało informacji o jej przebiegu oraz rokowaniu pacjentów.
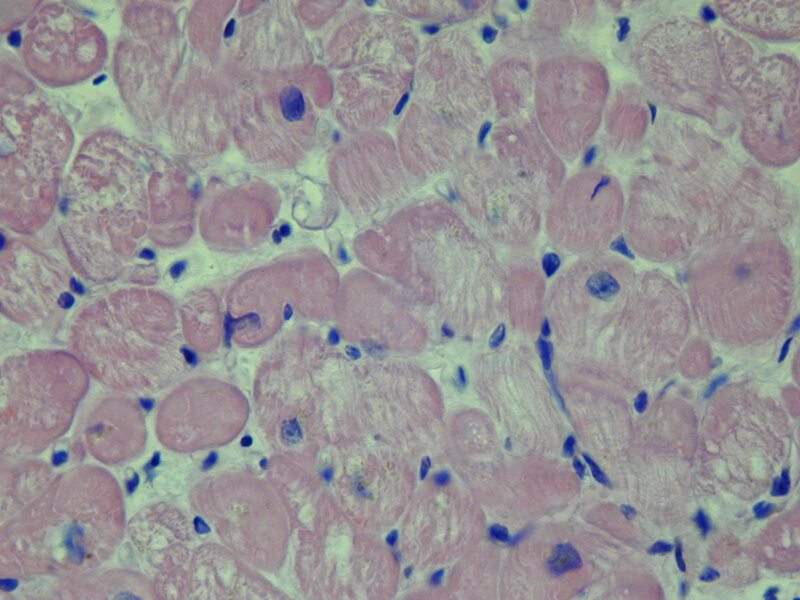
Rycina 13. Biopsja endomiokardialna. Nierozstrzeniowa kardiomiopatia lewej komory. Niespecyficzne zmiany wsteczne w kardiomiocytach w barwieniu hematoksyliną i eozyną
Podsumowanie
Pomimo rozwoju nowoczesnych technik diagnostycznych różnicowanie kardiomiopatii pozostaje wielkim wyzwaniem. Rozpoznanie kardiomiopatii jest przede wszystkim domeną badań klinicznych. Należy jednak podkreślić, że przyżyciowa diagnostyka morfologiczna serca wnosi dodatkowe, cenne informacje pozwalające ukierunkować postępowanie diagnostyczno-lecznicze. Dotyczy to w szczególności potwierdzenia lub wykluczenia podłoża zapalnego różnych schorzeń mięśnia sercowego. Ponadto biopsja mięśnia sercowego w wielu przypadkach pozwala na ostateczne ustalenie rozpoznania, tak jak w schorzeniach spichrzeniowych serca, co ma ogromne znaczenie w doborze odpowiedniej terapii.
Abstract
Clinical and morphological aspects of selected cardiomyopathies
Cardiomyopathies significantly contribute to the progression of heart failure. Despite their high incidence, the diagnosis of cardiomyopathies gives rise to considerable uncertainties and doubts both at the stage of identification of a given cardiomyopathy phenotype and during the diagnostic workup. The development of medicine in recent years has made it possible to expand and clarify the diagnostic panel. The extent of myocardial damage and morphological features of a given cardiomyopathy can be determined based on a carefully taken history and an echocardiogram, followed by an MRI examination and, in some cases, an endomyocardial biopsy. Although myocardial biopsy is an invasive procedure that involves some risk of complications, in many cases it is the only method that combined with clinical parameters, allows guiding the diagnosis and thus choosing an appropriate therapy for the patient.
- 1. Arbelo E, Protonotarios A, Gimeno JR, et al. Wytyczne ESC 2023 dotyczące postępowania w kardiomiopatiach. Polish Heart Journal (Kardiologia Polska) 2023;81(Supp. IV). Zeszyty Edukacyjne 2023;5-6
- 2. Cataldo P, Verdugo F, Appiani F, et al. Endomyocardial biopsy in current practice: Technical Aspects, Indications and complications. Rev Med Chil 2023;151(7):899-907. doi: 10.4067/s0034-98872023000700899
- 3. Biernacka EK, Mizia-Stec K. Kardiomiopatie. Warszawa: PZWL, 2024
- 4. Heymans S, Lakdawala NK, Tschöpe C, et al. Dilated cardiomyopathy: causes, mechanisms, and current and future treatment approaches. Lancet 2023;402(10406):998-1011. doi: 10.1016/S0140-6736(23)01241-2
- 5. Japp AG, Gulati A, Cook SA, et al. The Diagnosis and Evaluation of Dilated Cardiomyopathy. JACC 2016;67(25):2996-3010. doi: 10.1016/j.jacc.2016.03.590
- 6. Patel AR, Kramer CM. Role of Cardiac Magnetic Resonance in the Diagnosis and Prognosis of Non-Ischemic Cardiomyopathy. JACC Cardiovasc Imaging 2017;10(10 Pt A):1180-93. doi: 10.1016/j.jcmg.2017.08.005
- 7. Trachtenberg BH, Hare JM. Inflammatory cardiomyopathy syndromes. Circ Res 2017;121;803-18. doi: 10.1161/CIRCRESAHA.117.310221
- 8. Maisch B, Pankuweit S. Inflammatory dilated cardiomyopathy. Herz 2020;45(3):221-9. doi: 10.1007/s00059-020-04900-8
- 9. Kumar V, Abbas AK, Aster JC. Patologia Robbins. Wrocław: Edra Urban & Partner, 2019
- 10. Firth J. Cardiology: hypertrophic cardiomyopathy. Clin Med (Lond) 2019;19(1):61-3. doi: 10.7861/clinmedicine.19-1-61
- 11. Teekakirikul P, Zhu W, Huang HC, et al. Hypertrophic Cardiomyopathy: An Overview of Genetics and Management. Biomolecules 2019;9(12):878. doi: 10.3390/biom9120878
- 12. Haland TF, Edvardsen T. The role of echocardiography in management of hypertrophic cardiomyopathy. J Echocardiogr 2020;18(2):77-85. doi: 10.1007/s12574-019-00454-9
- 13. Hindieh W, Chan R, Rakowski H. Complementary Role of Echocardiography and Cardiac Magnetic Resonance in Hypertrophic Cardiomyopathy. Curr Cardiol Rep 2017;19(9):81. doi: 10.1007/s11886-017-0897-z
- 14. Adabag AS, Maron BJ, Appelbaum E, et al. Occurrence and frequency of arrhythmias in hypertrophic cardiomyopathy in relation to delayed enhancement on cardiovascular magnetic resonance. Am Coll Cardio 2008;51(14):1369-74. doi: 10.1016/j.jacc.2007.11.071
- 15. Hughes SE. The pathology of hypertrophic cardiomyopathy. Histopathology 2004;44(5):412-27. doi: 10.1111/j.1365-2559.2004.01835.x
- 16. Rapezzi C, Aimo A, Barison A, et al. Restrictive cardiomyopathy: definition and diagnosis. Eur Heart J 2022;43(45):4679-93. doi: 10.1093/eurheartj/ehac543
- 17. Muchtar E, Blauwet LA, Gertz MA. Restrictive Cardiomyopathy: Genetics, Pathogenesis, Clinical Manifestations, Diagnosis, and Therapy. Circ Res 2017;121(7):819-37. doi: 10.1161/CIRCRESAHA.117.310982
- 18. Ciarambino T, Menna G, Sansone G, et al. Cardiomyopathies: An Overview. Int J Mol Sci 2021;22(14):7722. doi: 10.3390/ijms22147722
- 19. Pilichou K, Thiene G, Bauce B, et al. Arrhythmogenic cardiomyopathy. Orphanet J Rare Dis 2016;11:33. doi: 10.1186/s13023-016-0407-1
- 20. Eda Y, Nabeta T, Iikura S, et al. Non-dilated left ventricular cardiomyopathy vs. dilated cardiomyopathy: clinical background and outcomes. ESC Heart Fail 2024;11(3):1463-71. doi: 10.1002/ehf2.14711
- 21. Tkaczyszyn M. Dilated versus non‐dilated left ventricular cardiomyopathy: Same same but different? ESC Heart Fail 2024;11(5):2681-3. doi: 10.1002/ehf2.14923
Następny artykuł:



 Dodaj do ulubionych
Dodaj do ulubionych
