Ponieważ u pacjentów z SLA mogą wystąpić objawy typowe dla bvFTD czy afazji pierwotnej postępującej (PPA – primary progressive aphasia), a z kolei u chorych z FTD obserwuje się niekiedy objawy choroby motoneuronu, podjęto kilka prób systematyzacji objawów zespołu, który w starszym piśmiennictwie nazywano FTD z chorobą neuronu ruchowego (FTD-MND – FTD with motor neuron disease) lub otępieniem w SLA (SLA-D). Najnowsze kryteria diagnostyczne systematyzujące spektrum SLA i FTD (SLA-FTSD – frontotemporal spectrum disorder of SLA) opracowane w 2017 r. przez Stronga i wsp.1 zostały przedstawione całościowo w tabeli 1. Nowe kryteria charakteryzują się wyższą czułością niż poprzednie, opracowane 2009 r.10 W nowych kryteriach diagnostycznych zoperacjonalizowano kryteria rozpoznawania zaburzeń poznawczych i zaburzeń zachowania typowych dla SLA oraz kryteria rozpoznawania otępienia (SLA-FTD). Wprowadzono też nową kategorię diagnostyczną, obejmującą zarówno zaburzenia poznawcze, jak i zaburzenia zachowania, ale bez otępienia (tab. 1).
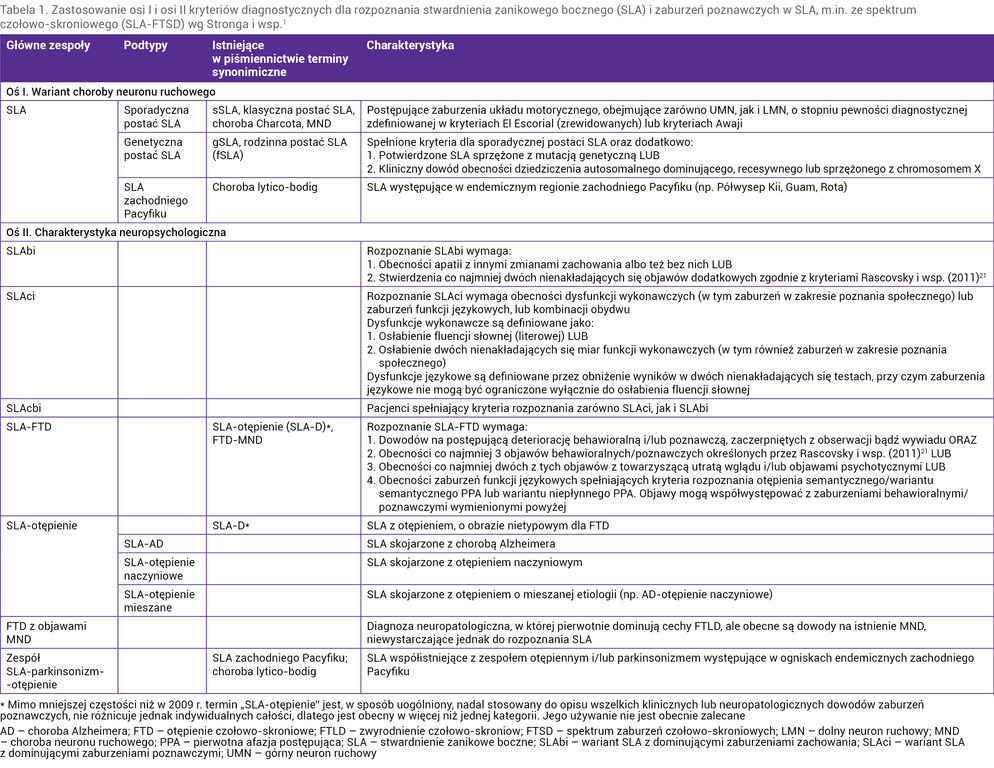
Tabela 1. Zastosowanie osi I i osi II kryteriów diagnostycznych dla rozpoznania stwardnienia zanikowego bocznego (SLA) i zaburzeń poznawczych w SLA, m.in. ze spektrum czołowo-skroniowego (SLA-FTSD) wg Stronga i wsp.1
Na podstawie włoskiej analizy danych klinicznych 797 pacjentów wykazano, że u 48,2% chorych nie stwierdzono zaburzeń poznawczych, 20,5% spełniało kryteria SLA-FTD, 4,8% miało zarówno zaburzenia poznawcze, jak i zaburzenia zachowania (SLAcbi – SLA with cognitive and behavioural impairment), 16,6% ujawniało izolowane zaburzenia poznawcze (SLAci – SLA with cognitive impairment), zaś 7,9% – izolowane zaburzenia zachowania (SLAbi – SLA with behavioral impairment). U 2% osób wykazano obecność zaburzeń poznawczych innych niż dysfunkcje wykonawcze11.
Badanie neuropsychologiczne pacjenta z SLA
W ocenie funkcji poznawczych chorego z SLA niezbędne jest uwzględnienie tych obszarów funkcjonowania poznawczego, w których spodziewamy się deficytów (ocena funkcji wykonawczych i poznania społecznego, pamięci operacyjnej i kompetencji językowej), oraz tych, w których zaburzenia obserwuje się w SLA rzadko (ocena pamięci epizodycznej, ocena funkcji wzrokowo-przestrzennych, praksji, jeśli sprawność motoryczna na to pozwala).
Dobór metod diagnostycznych wymaga uwzględnienia profilu objawów ruchowych i aktualnego stanu klinicznego pacjenta3. W przypadku niedowładu kończyn górnych zazwyczaj niemożliwe jest wykonanie prób wymagających reakcji motorycznej, co ogranicza zarówno metodologię oceny funkcji wzrokowo-przestrzennych (rezygnacja z oceny zdolności wzrokowo-konstrukcyjnych), jak i pamięci operacyjnej (odstąpienie od prób z ograniczeniem czasowym, wymagających reakcji motorycznej, takich jak Test Łączenia Punktów) oraz pamięci wzrokowej (rezygnacja z prób rysowania czy też układania wzorów). W przypadku występowania anartrii, ale bez znaczącego niedowładu, niekiedy pacjent udziela w badaniu odpowiedzi w formie pisemnej, wykorzystując pismo odręczne, pisanie na klawiaturze tradycyjnej lub z użyciem programu komputerowego, który śledzi ruchy gałek ocznych.
Najłatwiejszym sposobem udzielania odpowiedzi dla pacjenta z głębokimi zaburzeniami ruchowymi są testy wielokrotnego wyboru. Niewiele testów neuropsychologicznych jednak w formie oryginalnej zostało skonstruowanych w tym formacie. Przykładami takich przyjaznych dla pacjenta z SLA zadań są np. podtest Skojarzenia Semantyczne w Sydney Language Battery, oceniający pamięć semantyczną, Test Pamięci Krótkotrwałej Wzrokowej Bentona (BVRT) w wersjach F oraz G czy też Test Matryc Ravena, oceniający zdolność wnioskowania poprzez analogię na materiale wzrokowym.
W przypadku niektórych metod diagnostycznych możliwa jest ich adaptacja umożliwiająca pacjentowi udzielanie odpowiedzi w sposób, który będzie dla niego dostępny. Na przykład próba nazywania konfrontacyjnego może być przeprowadzona zarówno w formie ustnej, jak i pisemnej. Przykładem bardzo udanej adaptacji zadania dla pacjenta z SLA, co więcej, umożliwiającej zróżnicowanie wpływu czynników poznawczych i motorycznych na wynik, jest metodologia oceny fluencji słownej zaproponowana przez Sharon Abrahams i włączona do Edinburgh Cognitive and Behavioural ALS Screen (ECAS; https://ecas.psy.ed.ac.uk). Abrahams zaproponowała możliwość wykonania próby ustnie lub pisemnie, a następnie skorygowanie wyniku względem czasu wykonania próby odczytywania słów (wariant ustny) lub pisania pod dyktando (wariant pisemny). Pozwala to na uniezależnienie wyniku od tempa artykulacji lub pisania12.
Niektóre metody badania, na przykład próby uczenia się materiału słownego, są trudne do zaadaptowania dla osoby, która miałaby udzielać odpowiedzi pisemnej, gdyż w takiej formie wymieniany przez pacjenta pisemnie materiał pozostawałby w polu jego widzenia w toku odtwarzania, co mogłoby wpłynąć na wyniki. W przypadku krótkich prób uczenia (np. zapamiętywanie adresu z Addenbrooke’s Cognitive Examination III) jedna z autorek pracy wykorzystywała na przykład pisanie na klawiaturze odtwarzanych słów, ale z wykorzystaniem białej czcionki na białym tle. Taka procedura ma oczywiście charakter eksperymentu i nie wydaje się, aby mogła zastąpić tradycyjne sposoby odtwarzania w przypadku standardowych prób uczenia się materiału słownego.
Aby rozpoznać SLAci u pacjenta z diagnozą SLA, niezbędne jest wykazanie deficytów wykonawczych lub językowych. W przypadku dysfunkcji wykonawczych można je udokumentować na podstawie słabego wyniku próby fluencji literowej lub też słabych wyników dwóch niezależnych prób oceniających funkcje wykonawcze. Analogicznie, w przypadku istotnych zaburzeń językowych ich potwierdzeniem są wyniki dwóch niezależnych testów oceniających te funkcje. Wybiórcze obniżenie fluencji słownej nie stanowi podstawy do rozpoznania zaburzeń językowych w SLA, gdyż może wynikać z deficytów wykonawczych. Na przykład aby wykazać zaburzenia językowe w SLA, zgodnie z kryteriami diagnostycznymi, można by zastosować test nazywania i skojarzeń semantycznych (taki jak z Sydney Language Battery).
W przyszłości być może szersze wykorzystanie technologii śledzenia ruchu gałek ocznych umożliwi wprowadzenie do powszechnej praktyki klinicznej metod oceny, które nie będą wymagały od pacjenta mówienia ani pisania z zaangażowaniem motoryki3,13.
Najczęściej zalecanym narzędziem przesiewowej oceny funkcji poznawczych w SLA jest wspomniana już powyżej skala ECAS. Zasadniczo takie narzędzie nie powinno zawierać prób wymagających reakcji motorycznej4. W przypadku diagnozy pacjenta z mniej nasilonymi objawami zajęcia dolnego neuronu ruchowego i braku dostępu do neuropsychologa niekiedy rekomendowane jest wykorzystanie skali Frontal Assessment Battery (FAB), która pozwala na przesiewową ocenę w kierunku deficytów wykonawczych3.
Ocena zaburzeń zachowania w SLA
Istnieje wiele skal opracowanych pod kątem wykrywania zaburzeń zachowania w SLA, ale żadna z nich nie jest powszechnie wykorzystywana w praktyce klinicznej. Przeprowadzając wywiad w kierunku zaburzeń zachowania z pacjentem i wiarygodnym informatorem, należy zwrócić szczególną uwagę na objawy bvFTD z kryteriów diagnostycznych oraz na symptomy psychotyczne. Dodatkowo w wywiadzie warto uwzględnić zmianę wrażliwości na bodźce bólowe i nadwrażliwość sensoryczną, spadek krytycyzmu, drażliwość, bladość afektywną, zachowania perseweracyjne (zarówno proste, jak i złożone), zbieractwo, echolalię i zachowanie użytkownika (utilization behaviour; zachowanie wskazujące na zależność od otoczenia, które nie jest inicjowane w sposób wolicjonalny, ale wywołane bodźcem pojawiającym się w zasięgu wzroku/ręki osoby chorej, np. odczytuje ona treść plakietki lekarza czy napis na ścianie, bierze do ręki długopis i zaczyna pisać, sięga po filiżankę i zaczyna pić, nawet jeśli dopiero się napiła i nie jest to jej filiżanka; objaw ten wskazuje na znaczący deficyt hamowania). Okresem porównawczym dla zmiany zachowania może być 10 lat przed pojawieniem się objawów ruchowych, tak jak w Beaumont Behavioural Questionnaire, który jest narzędziem kompleksowo oceniającym potencjalne zmiany zachowania w SLA14.
Znaczenie badania genetycznego w diagnostyce różnicowej SLA-FTSD
W ostatnich latach dokonał się znaczny postęp w zrozumieniu podłoża genetycznego SLA i FTD. Nakładające się fenotypy obu chorób związane są szczególnie z mutacjami w genach C9orf72, SQSTM1, VCP, FUS, TBK1. W Europie najczęstszą przyczyną rodzinnej postaci SLA (fSLA – familial SLA) i rodzinnej FTD (fFTD – familial FTD) jest zwielokrotnienie powtórzeń w genie C9orf72 odpowiadające za ok. 40% fSLA i 25% fFTD15. Związane z tą mutacją SLA-FTD dziedziczy się autosomalnie dominująco, jednak ma ona niepełną penetrację. Niektóre mutacje związane są przede wszystkim z SLA i rzadko występują w FTD. Są to SOD1, FUS i warianty TARDBP. Podobnie mutacje w genie dla progranuliny częste w FTD nie występują w SLA16.
Wiedza na temat podłoża genetycznego obu chorób może być pomocna przy próbie odpowiedzi na tak często zadawane przez pacjentów pytania: dlaczego ja, czy moje dzieci są także zagrożone chorobą, jak szybki będzie postęp choroby? Diagnostyka genetyczna jest jednak trudno dostępna mimo zapotrzebowania ze strony pacjentów i lekarzy. Analiza zachorowań na SLA, FTD, inne otępienia, choroby psychiatryczne, parkinsonizm czy samobójstw w rodzinie (co najmniej trzech pokoleń) może sugerować sposób dziedziczenia choroby, na przykład transmisja autosomalna dominująca jest stosunkowo częsta w SLA. Niestety ograniczona penetracja genu, brak informacji o rodzinie, błędne rozpoznania, wczesny zgon, nieujawniona adopcja i inne okoliczności mogą doprowadzić do błędnych wniosków. Prawdopodobieństwo nosicielstwa znanej mutacji o wysokiej penetracji w przypadku sporadycznego zachorowania na SLA wynosi 11%. Natomiast przy fenotypie SLA-FTD wynosi już 88% nawet przy ujemnym wywiadzie rodzinnym. W obu przypadkach najczęstszą mutacją jest zwielokrotnienie powtórzeń w genie C9orf7217. Wszyscy pacjenci z SLA, którzy mają europejskie korzenie, powinni mieć możliwość przeprowadzenia badania w kierunku mutacji w C9orf72, ponieważ dostępne są już badania kliniczne dla nosicieli tej mutacji. Badania u osób zdrowych powinny być zarezerwowane dla dorosłych krewnych pierwszego stopnia z potwierdzoną mutacją.
Znaczenie badań obrazowych w diagnostyce SLA-FTSD
Badania obrazowe w diagnostyce SLA-FTSD mają charakter pomocniczy i, w przeciwieństwie do bvFTD czy PPA, żaden ze wzorców wyników badań neuroobrazowych nie zwiększa pewności rozpoznania. Badania obejmujące ocenę morfologiczną z zastosowaniem rezonansu magnetycznego: morfometria oparta na wokselach (VBM – voxel-based morphometry) i morfometria oparta na powierzchni (SBM – surface-based morphometry), wykorzystując wysokiej rozdzielczości MR, pozwalają na wykrywanie ogniskowych zmian w obrębie istoty szarej. Technika ta pozwala (poza uwidacznianiem zmian w korze ruchowej, co do których nie ma wątpliwości, biorąc pod uwagę patomechanizm choroby) również na zobrazowanie wieloogniskowych zmian w okolicach czołowej, skroniowej i ciemieniowej18. Zmiany ogniskowe opisywane były też w strukturach podkorowych, w tym w hipokampie, ciele migdałowatym, wzgórzu i wyspie19. Rozmieszczenie zmian korowych w strukturach pozaruchowych potwierdza kontinuum SLA-FTSD.