Choroby autozapalne u dzieci
prof. dr hab. n. med. Elżbieta Smolewska

prof. dr hab. n. med. Elżbieta Smolewska
- Charakterystyka obrazu klinicznego i przebiegu chorób autozapalnych w populacji pediatrycznej
- Diagnostyka wrodzonych zespołów gorączek nawrotowych – kryteria Eurofever/PRINTO z 2019 roku
- Zastosowanie leków biologicznych w leczeniu chorób autozapalnych
Choroby autozapalne stanowią stosunkowo nową grupę chorób, funkcjonującą oficjalnie w medycynie od 1997 roku, kiedy to został zidentyfikowany gen odpowiedzialny za występowanie rodzinnej gorączki śródziemnomorskiej (FMF – familial Mediterranean fever). Pod koniec lat 90. XX wieku opisano dwie inne autozapalne jednostki chorobowe: zespół hiperimmunoglobulinemii D (HIDS – hyperimmunoglobulin D syndrome) oraz zespół gorączki nawrotowej zależny od receptora czynnika martwicy nowotworów (TRAPS – tumor necrosis factor [TNF] receptor-associated periodic syndrome). W ciągu ostatnich 20 lat opisano kolejne zespoły chorobowe o podłożu autozapalnym charakteryzujące się dużą heterogennością i na ogół wczesnym początkiem objawów1. Patofizjologię procesu autozapalnego cechują przewlekły przebieg oraz uszkodzenia o długoterminowym charakterze. Od kilku lat dostępne są leki skutecznie kontrolujące mechanizmy autozapalenia oraz główne objawy, co umożliwia pacjentom dotkniętym tymi genetycznie uwarunkowanymi chorobami zachowanie akceptowalnej jakości życia, chociaż wciąż nie ma skutecznej terapii większości ciężkich manifestacji. Na ogół leczenie farmakologiczne jest oparte na grupie leków biologicznych blokujących cząsteczki zaangażowane w proces zapalny, które są produkowane w nasilonej ilości w ostrej fazie epifenomenu zapalnego2.
Definicja, obraz kliniczny i patofizjologia chorób autozapalnych
Choroby autozapalne stanowią grupę chorób, które ujawniają się w wyniku dysfunkcji lub dysregulacji układu odporności wrodzonej i mogą powodować poważne powikłania oraz ryzyko zgonu poprzez zajęcie wielu narządów. Ta nowa grupa chorób różni się od standardowej koncepcji chorób z autoimmunizacji, które są relatywnie dobrze poznane w zakresie podstawowych mechanizmów, defektem odporności wrodzonej i brakiem krążących autoprzeciwciał w krwiobiegu. Termin „choroby autozapalne” został po raz pierwszy użyty przez amerykańskie National Institutes of Health na określenie systemowego zapalenia charakteryzującego się nieprowokowanymi atakami, bez obecności przeciwciał i antygenowo swoistych limfocytów T. Z kolei Kastneri i wsp. w 2010 roku zdefiniowali choroby autozapalne jako choroby cechujące się nadmiernie nasilonym zapaleniem mediowanym głównie przez komórki i cząsteczki układu odporności wrodzonej u osób predysponowanych. W 2017 roku Wekell i wsp. zmodyfikowali tę definicję, określając choroby autozapalne jako: „choroby immunologicznie uwarunkowane z nadmiernym zapaleniem spowodowanym przez dysregulację komórek i cząsteczek wrodzonej odporności, przy znacznej predyspozycji gospodarza, ale często również z aktywacją układu odporności nabytej i dysfunkcją immunologiczną (podatność na infekcje), autoimmunizacją lub niekontrolowanym nadmiernym zapaleniem”. Ostateczna definicja chorób autozapalnych została sformułowana przez Paediatric Rheumatology INternational Trials Organisation (PRINTO) w 2018 roku, zgodnie z którą to „jednostki kliniczne spowodowane defektem bądź dysregulacją wrodzonej odpowiedzi immunologicznej, charakteryzujące się nawracającym lub ciągłym zapaleniem (podwyższone wskaźniki ostrej fazy) i brakiem pierwotnej roli patogennej układu odporności nabytej (brak autoreaktywnych limfocytów T lub produkcji autoprzeciwciał)”.
Choroby autozapalne mają różny obraz kliniczny w zależności od specyficznej mutacji genetycznej odpowiedzialnej za chorobę i cechują się występowaniem zapalenia powstałego w wyniku uwalniania specyficznych cytokin prozapalnych (głównie interleukiny [IL] 1, TNFα czy interferonów [IFN] α, β). Początek objawów chorobowych obserwuje się już w pierwszych miesiącach/latach życia z gorączką, zajęciem stawów, skóry oraz obecnością markerów serologicznych. Objawy choroby autozapalnej pojawiają się nagle, w stanie zdrowia, z charakterystyczną wysoką ciepłotą ciała (do 39-40°C)3,4.
Klasyfikacja chorób autozapalnych ze względu na patogenezę
Podział chorób autozapalnych w zależności od czynnika patofizjologicznego przedstawiono w tabeli 1.
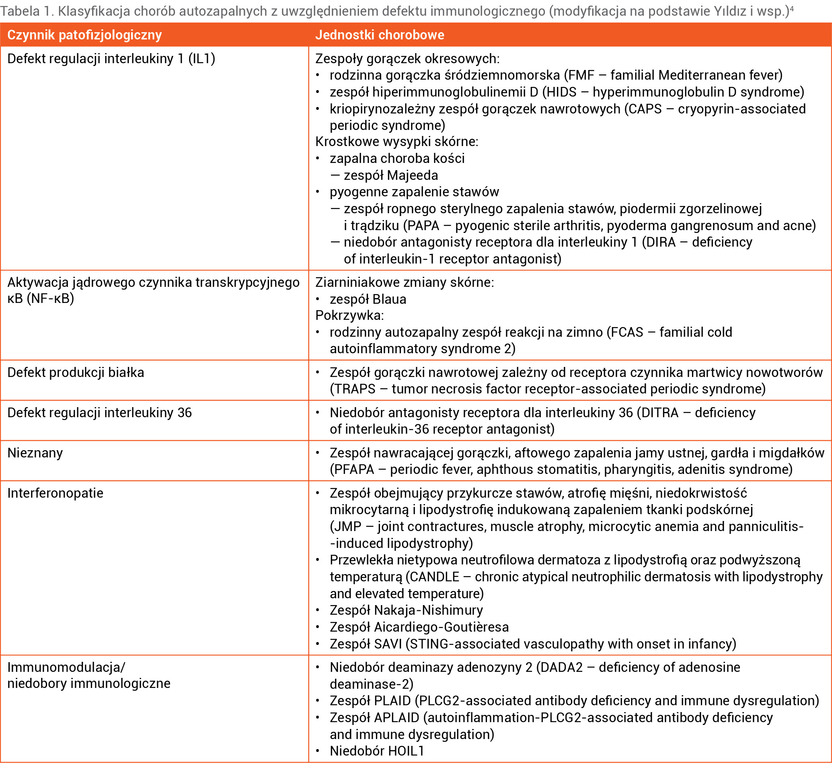
Tabela 1. Klasyfikacja chorób autozapalnych z uwzględnieniem defektu immunologicznego (modyfikacja na podstawie Yıldız i wsp.)4
Zespoły gorączek nawrotowych
Zespoły gorączek nawrotowych (periodic fever syndromes) to heterogenna grupa układowych chorób autozapalnych, które charakteryzują się nawracającymi epizodami zapalenia bez związku z infekcją, procesem autoimmunizacyjnym czy z chorobą nowotworową. U podłoża większości chorób z tej grupy leżą mutacje genów kodujących białka związane ze szlakami reakcji zapalnej, apoptozą i wytwarzaniem cytokin, prowadzące do zaburzenia mechanizmów odporności wrodzonej. Większość chorób z tej grupy ma początek w wieku rozwojowym. Dominującym objawem są epizody gorączki, której towarzyszą zmiany skórne, zapalenie stawów, dolegliwości ze strony przewodu pokarmowego (ból brzucha, biegunka). W czasie zaostrzenia choroby stwierdza się istotnie zwiększone wartości wskaźników stanu zapalnego oraz stężenia cytokin prozapalnych. W okresie między rzutami objawy ustępują, a wyniki badań laboratoryjnych zazwyczaj się normalizują.
Zespoły gorączek nawrotowych są wyzwaniem diagnostycznym dla klinicysty. Kluczowe jest przeprowadzenie szerokiej diagnostyki różnicowej uwzględniającej infekcje, choroby z autoimmunizacji czy rozrostowe. Przed ustaleniem rozpoznania wskazane są obserwacja pacjenta przez minimum 6 miesięcy, notowanie czasu trwania gorączki i okresów bezobjawowych, udokumentowanie wzrostu wartości wskaźników stanu zapalnego w momencie rzutu choroby. Istotne jest dokładne badanie podmiotowe pacjenta, uwzględniające wiek ujawnienia się objawów, pochodzenie etniczne chorego oraz występowanie podobnych objawów w rodzinie.
Do najczęściej rozpoznawanych zespołów gorączek nawrotowych należą:
- rodzinna gorączka śródziemnomorska (FMF)
- zespół gorączki nawrotowej zależny od receptora dla TNF (TRAPS)
- zespół hiperimmunoglobulinemii D (HIDS)
- kriopirynozależny zespół gorączek nawrotowych (CAPS)
- zespół nawracającej gorączki, aftowego zapalenia jamy ustnej, gardła i migdałków (PFAPA).
Rodzinna gorączka śródziemnomorska
Jest najczęstszą monogenową chorobą autozapalną na świecie. Największą liczbę zachorowań odnotowuje się w basenie Morza Śródziemnego, tj. wśród Arabów, Turków, Ormian, Żydów sefardyjskich. W innych populacjach FMF należy do rzadkości. U większości chorych pierwsze objawy pojawiają się przed 10 rokiem życia. Choroba jest dziedziczona autosomalnie recesywnie. U podłoża FMF leżą mutacje genu MEFV kodującego pirynę. Zmieniona cząsteczka tego białka staje się nieefektywnym inhibitorem odpowiedzi zapalnej, co prowadzi do nasilenia apoptozy i produkcji interleukiny 1β (IL1β). Rzut choroby trwa zwykle 1-4 dni, często jest poprzedzony infekcją, urazem lub stresem. Wysokiej gorączce towarzyszy zapalenie błon surowiczych, najczęściej otrzewnej, mogące imitować objawy tzw. ostrego brzucha. Rzadziej występują: zapalenie opłucnej (objawiające się zazwyczaj bólem w klatce piersiowej), osierdzia, moszny (objawy sugerujące skręt jądra), zapalenie opon mózgowo-rdzeniowych. U blisko 50% chorych stwierdza się przemijające zapalenie dużych stawów (najczęściej kolanowego, biodrowego, skokowego). U 25% pacjentów na grzbietowej powierzchni stóp i stawu skokowego pojawia się przemijająca, różopodobna wysypka. W początkowym okresie FMF możliwe jest występowanie gorączki jako izolowanego objawu choroby.
Rozpoznanie FMF ustala się na podstawie nowych kryteriów klinicznych Eurofever/PRINTO, które obejmują stwierdzenie mutacji w genie MEFV oraz spełnienie co najmniej jednego z 4 następujących kryteriów: rzuty choroby trwające 1-3 dni, zapalenie stawów, ból w klatce piersiowej, ból brzucha. W przypadku braku mutacji patognomonicznych w genie MEFV wskazane jest spełnienie minimum 2 z powyższych kryteriów.