



Co znajdziesz w artykule?
- Twardzina układowa – problem interdyscyplinarny, wymagający współpracy dermatologa, pulmonologa i reumatologa
- Przebieg choroby i możliwe powikłania, z naciskiem na problemy pulmonologiczne
- Nowoczesne metody terapeutyczne
Spis treści
- Epidemiologia i obraz kliniczny twardziny układowej
- Rozpoznanie twardziny układowej
- Śródmiąższowa choroba płuc w przebiegu twardziny układowej
- Badania przeciwciał (panel reumatologiczny) w SSc-ILD
- Badania radiologiczne w SSc-ILD
- Badania czynnościowe układu oddechowego w SSc-ILD
- Możliwości terapeutyczne w SSc
- Zasady postępowania w SSc-ILD
- Koncepcja postępującego włóknienia płuc
- Podsumowanie
Epidemiologia i obraz kliniczny twardziny układowej
Twardzina układowa (SSc – systemic sclerosis) jest przewlekłą układową chorobą tkanki łącznej o podłożu autoimmunologicznym. W jej przebiegu dochodzi do mikroangiopatii, tworzenia specyficznych autoprzeciwciał oraz ostatecznie włóknienia skóry i narządów wewnętrznych. Najistotniejsze z punktu widzenia jakości życia pacjenta i rokowania są włóknienie i owrzodzenia skóry, zajęcie płuc (śródmiąższowa choroba płuc [ILD – interstitial lung
disease], tętnicze nadciśnienie płucne [PAH – pulmonary arterial hypertension]) oraz uszkodzenie struktur układu ruchu (włóknienie ścięgien i więzadeł, zapalenie i ograniczenie ruchomości stawów przez stwardniałą skórę). Stąd wynika podstawowa rola i potrzeba bliskiej współpracy dermatologa, pulmonologa i reumatologa w opiece nad chorym z SSc.
Patomechanizm choroby nie jest w pełni poznany, jednak podkreśla się udział 3 zasadniczych mechanizmów – przewlekłego uszkodzenia mikrokrążenia, aktywacji endotelium oraz układu immunologicznego, które prowadzą do pobudzenia fibroblastów i wtórnego włóknienia narządów.
Na całym świecie SSc rozpoznaje się u blisko 2,5 mln osób 1 . W Europie i Ameryce Północnej częstość jej występowania wynosi odpowiednio 7,2-33,9 i 13,5-44,3 na 100 000 osób 2 . Jednocześnie u ok. 35% Europejczyków i ok. 52% Amerykanów stwierdza się współistnienie ILD. Dziesięcioletnie przeżycie ocenia się na 65-73% w Europie i 54-82% w Ameryce Północnej. W Polsce, wg danych Narodowego Funduszu Zdrowia (NFZ), na SSc choruje ok. 54 000-59 000 osób 3, 4 .
Większość chorych na SSc stanowią kobiety, które zapadają na nią 3-4-krotnie częściej niż mężczyźni 1 . Choroba może wystąpić w każdym wieku, ale najczęściej jest rozpoznawana u osób w wieku 25-55 lat 1 .
SSc jest zaliczana do chorób rzadkich i (mimo postępu medycyny) cechujących się złym rokowaniem. Obecnie główną przyczyną zgonów w przebiegu SSc są powikłania ze strony układu oddechowego – ILD i PAH – stanowiące 60% wszystkich przyczyn śmierci w tej grupie chorych. Pomimo rozwoju medycyny odsetek zgonów z powodów pulmonologicznych w ostatniej dekadzie wzrósł, przy jednoczesnej znacznej redukcji śmiertelności związanej z niewydolnością nerek (z 42% do 6%), chorobami przewodu pokarmowego (z 12% do 4%) czy powikłaniami kardiologicznymi (z 10% do 5%) 5 .
Pomimo rzadkiego występowania, SSc stanowi znaczne obciążenie społeczne i ekonomiczne, szczególnie w postaci z zajęciem płuc (SSc-ILD). Roczny koszt opieki nad pacjentem z rozpoznaniem SSc-ILD zawiera się w przedziale od 6191 (Grecja) do 25 354 (Szwecja) euro, co ma związek przede wszystkim z częstymi hospitalizacjami 6 . Szacuje się również, że 40,4% pacjentów z SSc wymaga wcześniejszych (średnio o ok. 12 lat) świadczeń rentowych.
Wyróżnia się 3 zasadnicze postacie SSc, różniące się obrazem klinicznym, występowaniem przeciwciał oraz dynamiką uszkodzeń narządowych (tab. 1). Podstawowe znaczenie ma wczesne ustalenie podtypu choroby, ponieważ określa on zarówno jej przebieg (a co za tym idzie – diagnostykę i leczenie), jak i determinuje rokowanie. W opiece nad pacjentem z SSc należy szczególnie pamiętać o kilku istotnych prognostycznie sytuacjach klinicznych 8 :

Tabela 1. Obraz kliniczny twardziny układowej7
- Zespół dużego ryzyka rozwoju SSc (występowanie objawu Raynauda, typowych zmian w kapilaroskopii oraz swoistych autoprzeciwciał, bez zmian skórnych i narządowych) – rozwój choroby następuje w ciągu 5 lat u 65-80% pacjentów.
- Szybkie narastanie duszności i/lub prawokomorowej niewydolności serca u pacjenta z wieloletnim wywiadem SSc – rozwój PAH.
- Zmiany narządowe w pierwszych 3 latach postaci uogólnionej SSc – określenie dalszego przebiegu choroby i rokowania.
- Pojawienie się nadciśnienia tętniczego i białkomoczu (szczególnie u mężczyzny z wysiękowym zapaleniem osierdzia, leczonego glikokortykosteroidami lub z obecnością przeciwciał przeciwko RNA polimerazie III) – zagrożenie twardzinowym przełomem nerkowym.
Rozpoznanie twardziny układowej
Rozpoznanie późnej SSc nie stanowi zwykle problemu ze względu na typowe twardzinowe zmiany skórne oraz objawy niewydolności narządowej. Początkowo skóra ulega obrzękowi, następnie przechodzi fazę twardnienia, a później zaniku. W skórze mogą powstawać obszary hiperpigmentacji, teleangiektazje i zwapnienia. W postaci ograniczonej choroby często dochodzi do powstawania bolesnych, trudno gojących się owrzodzeń rąk i stóp, zaniku opuszek palców, paznokci i skrócenia paliczków dystalnych. Z czasem zmienia się wyraz twarzy pacjenta – staje się ona maskowata, z napiętą skórą oraz wąskimi ustami otoczonymi promienistym bruzdowaniem. W skrajnych przypadkach dochodzi do znacznego ograniczenia możliwości otwarcia ust.
Ocena zmian skórnych uzupełniona o badania serologiczne (oznaczanie autoprzeciwciał) stanowi podstawę obowiązujących od 2013 r. kryteriów klasyfikacyjnych (wykorzystywanych również diagnostycznie), przyjętych zarówno przez EULAR (European Alliance of Associations for Rheumatology), jak i ACR (American College of Rheumatology) 9 . Kryteria te, z uwzględnieniem domeny skórnej, naczyniowej i serologicznej, przedstawiono w tabeli 2. Warto zwrócić uwagę, że stwierdzenie typowych zmian skórnych – stwardnienia skóry obu rąk proksymalnie od stawów śródręczno-paliczkowych – pozwala na rozpoznanie SSc nawet bez wykazania obecności autoprzeciwciał w surowicy. Jednocześnie należy podkreślić, że najnowsze kryteria klasyfikacyjne umożliwiają postawienie rozpoznania już we wczesnym okresie choroby.
Tabela 2. Kryteria klasyfikacyjne twardziny układowej (na podstawie ACR/EULAR 2013)9
Rozpoznanie wczesnej SSc pozostaje nadal dużym wyzwaniem dla klinicysty. Ma ono jednak kluczowe znaczenie dla poprawy rokowania, ponieważ wydaje się, że odpowiednio szybkie włączenie leczenia może zapobiec powstaniu zagrażających życiu powikłań narządowych. Grupa EUSTAR (EULAR Scleroderma Trials and Research) zaproponowała algorytm postępowania w przypadku podejrzenia bardzo wczesnej postaci SSc (very early SSc) 10 . Podkreśla się w nim dużą wartość diagnostyczną objawu Raynauda (wyprzedza wystąpienie SSc nawet o kilka lat) oraz obrzęku palców rąk (puffy fingers). Każdego pacjenta, u którego stwierdza się oba wymienione objawy, należy skierować na badanie kapilaroskopowe oraz oznaczenie przeciwciał przeciwjądrowych (ANA – antinuclear antibodies). Obecność typowych zmian w kapilaroskopii – megakapilar (częściej w ograniczonej postaci twardziny) i obszarów pozbawionych naczyń (częściej w postaci uogólnionej) lub (!) typowych przeciwciał (przeciwko topoizomerazie I, antycentromerowych, przeciwko RNA polimerazie III) nakazuje skierowanie pacjenta na badania z zakresu diagnostyki narządowej. Jeśli w badaniach obrazowych lub czynnościowych oceniających układ oddechowy, sercowo-naczyniowy i pokarmowy stwierdzone zostaną typowe dla SSc odchylenia, to pomimo braku zmian skórnych można rozpoznać tę chorobę i rozpocząć terapię.
Śródmiąższowa choroba płuc w przebiegu twardziny układowej
Wystąpienie śródmiąższowej choroby płuc w przebiegu twardziny układowej (SSc-ILD) może dotyczyć nawet 90% chorych, jednak większość rozpoznań ustala się autopsyjnie 11 . Pacjenci, u których dochodzi do rozwoju ILD, to zwykle młodzi ludzie: 50% z nich ma mniej niż 50 lat. U 1 na 4 osoby z rozpoznaną SSc w ciągu 3 lat od postawienia diagnozy rozwija się ILD 12 . Zgodnie z danymi NFZ w 2019 r. w Polsce żyło nieco ponad 1700 osób z rozpoznaną ILD w przebiegu SSc 3 .
W wielu przypadkach zajęcia płuc objawy mogą być nieobecne lub bardzo skąpe. Pacjenci zwykle zgłaszają się do lekarza, kiedy zaczynają odczuwać osłabienie, postępującą duszność podczas wysiłku, męczący, suchy kaszel. Wraz z czasem trwania SSc-ILD postępuje włóknienie płuc, dochodzi do niewydolności oddechowej i inwalidztwa oddechowego. Z uwagi na początkowe skąpe objawy kliniczne zaleca się, by u chorych na SSc okresowo wykonywać ocenę układu oddechowego w celu wczesnego wykrycia ILD.
W badaniu przedmiotowym u części pacjentów można stwierdzić trzeszczenia, które zwykle w początkowym okresie obejmują dolne pola płucne, ale z czasem mogą się rozprzestrzeniać ku górze. W przypadku zaawansowanej choroby można wykazać tachypnoe, pracę dodatkowych mięśni oddechowych oraz tachykardię.
Badania przeciwciał (panel reumatologiczny) w SSc-ILD
Oznaczanie profilu ANA ma istotne znaczenie dla rozpoznania SSc. Pozwala również zaplanować terapię oraz monitorować postęp choroby, ze szczególnym uwzględnieniem zmian narządowych. Najczęściej występujące przeciwciała wykrywane w surowicy pacjentów z SSc, a także ich znaczenie kliniczne przedstawiono w tabeli 3 11 .
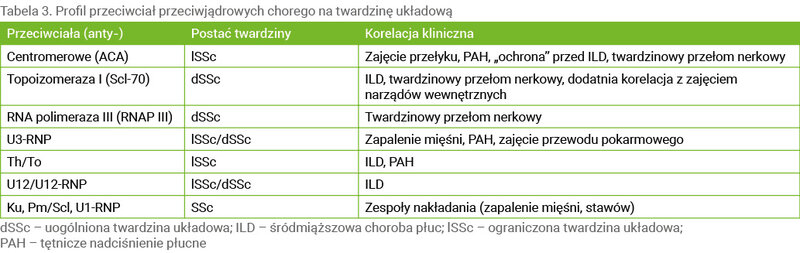
Tabela 3. Profil przeciwciał przeciwjądrowych chorego na twardzinę układową
Badania radiologiczne w SSc-ILD
W diagnostyce SSc-ILD złotym standardem jest wykonanie tomografii komputerowej wysokiej rozdzielczości (HRCT – high resolution computed tomography) klatki piersiowej. Badanie przeprowadza się bez podania środka kontrastowego. Pozwala ono z dużą czułością stwierdzić obecność zmian śródmiąższowych w płucach oraz określić ich charakter. Zasadnicze znaczenie ma ocena morfologii zmian – czy w obrazie dominuje aktywne zapalenie (obraz matowej szyby, histopatologiczny obraz niespecyficznego śródmiąższowego zapalenia płuc [NSIP – non-specific interstitial pneumonia] – częściej), czy śródmiąższowe włóknienie (wzorzec zwykłego śródmiąższowego zapalenia płuc [UIP – usual interstitial pneumonia]) (ryc. 1-4). HRCT często ujawnia nieprawidłowości śródmiąższowe w płucach nawet u pacjentów z prawidłowymi wynikami badań czynnościowych płuc lub bez objawów.
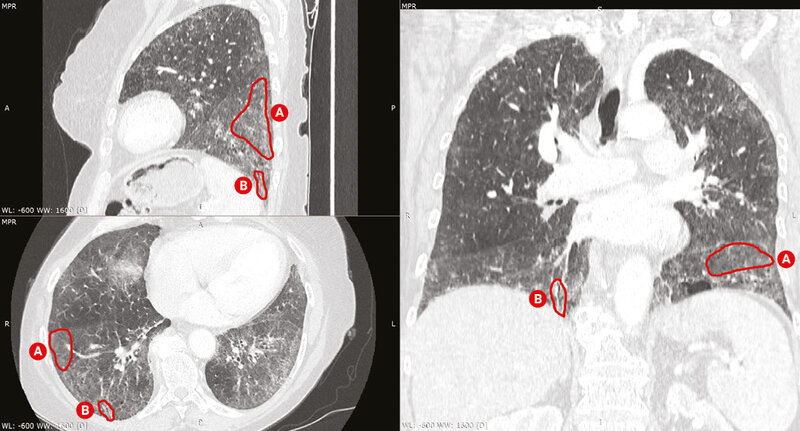
Rycina 1. Skany tomografii komputerowej klatki piersiowej w rekonstrukcji wielopłaszczyznowej u chorej z twardziną układową. Obraz nieswoistego śródmiąższowego zapalenia płuc z dominującym obrazem matowej szyby. Widoczne obszary matowej szyby (A) oraz pojedyncze rozstrzenie oskrzeli/oskrzelików z pociągania (B)
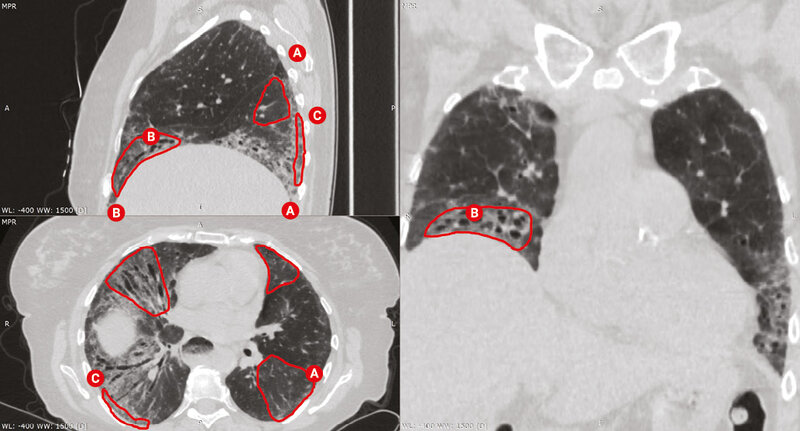
Rycina 2. Skany tomografii komputerowej klatki piersiowej w rekonstrukcji wielopłaszczyznowej u chorej z twardziną układową. Obraz włókniejącej postaci nieswoistego śródmiąższowego zapalenia płuc z widocznymi obszarami matowej szyby (A), siateczki i centralnych rozstrzeni oskrzeli z pociągania (B). Zlokalizowany podopłucnowo, zaoszczędzony rąbek miąższu płucnego (C)
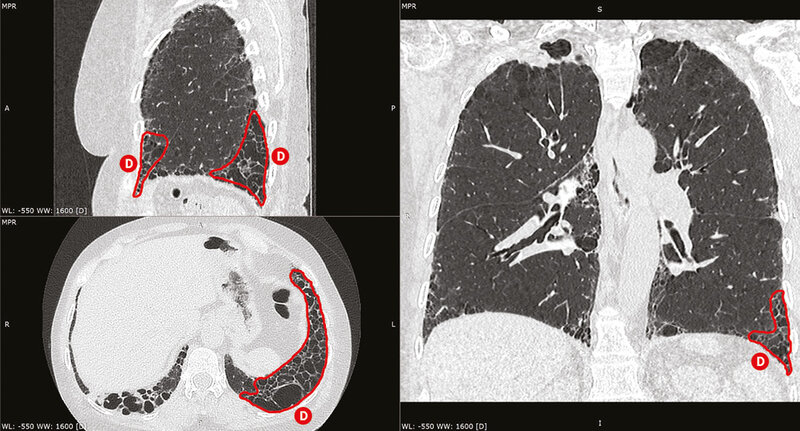
Rycina 3. Skany tomografii komputerowej klatki piersiowej w rekonstrukcji wielopłaszczyznowej u chorej z twardziną układową. Obraz włóknienia płuc o typie zwykłego śródmiąższowego zapalenia płuc (UIP). Dominuje plaster miodu zlokalizowany podopłucnowo (D)
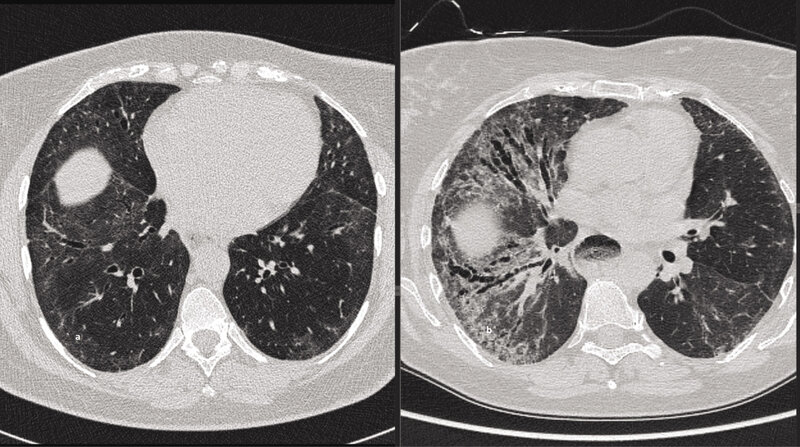
Rycina 4. Dwa skany tomografii komputerowej klatki piersiowej (HRCT) w przekroju poprzecznym na podobnym poziomie u chorej z twardziną układową (wykonane w odstępie 7 lat). Widoczna istotna progresja śródmiąższowego włóknienia. W celu wykazania progresji choroby śródmiąższowej płuc i postępującego włóknienia: po stronie lewej mało zaawansowane zmiany o typie matowej szyby, po stronie prawej cechy nasilonego śródmiąższowego włóknienia z widocznymi rozstrzeniami oskrzeli z pociągania
Badanie RTG klatki piersiowej charakteryzuje się zbyt małą czułością, prawidłowy obraz nie wyklucza zajęcia śródmiąższu płuc. Z tego powodu badanie to nie jest zalecane w diagnostyce choroby śródmiąższowej.
Badania czynnościowe układu oddechowego w SSc-ILD
Badania czynnościowe płuc powinny być wykonywane u pacjentów z SSc w momencie rozpoznania choroby, w celu oceny czynności płuc. Następnie wykonuje się je zwykle raz w roku lub wcześniej, jeśli stwierdzi się u chorego duszność, kaszel lub nieprawidłowości radiologiczne.
W skład oceny czynnościowej układu oddechowego wchodzą:
- Spirometria – szeroki dostęp do spirometrów spowodował, że najbardziej uznanym rokowniczo parametrem czynnościowym jest roczna utrata natężonej pojemności życiowej płuc (FVC – forced vital capacity). Proporcjonalne zmniejszenie FVC i pierwszosekundowej natężonej objętości wydechowej (FEV1 – forced expiratory volume in one second) sugeruje zaburzenia typu restrykcyjnego i może występować u pacjentów z ILD. Jednak prawidłowy wynik spirometrii nie wyklucza wczesnej postaci ILD w przebiegu SSc. Jedno z badań wykazało, że 62% pacjentów z SSc z prawidłową FVC miało klinicznie istotną ILD zidentyfikowaną w HRCT 13 .
- Pletyzmografia – ocena całkowitej pojemności płuc (TLC – total lung capacity) jest przeprowadzana w celu potwierdzenia restrykcji.
- Ocena wskaźnika transferu płucnego dla tlenku węgla (TLCO – transfer factor of the lung for carbon monoxide) – obniżenie TLCO może być pierwszą nieprawidłowością obserwowaną w ILD związanej z SSc, ale jest niespecyficzne; inne procesy (np. nadciśnienie płucne, palenie papierosów, choroba zakrzepowo-zatorowa) również mogą powodować obniżenie TLCO. Wśród pacjentów z ILD związaną z SSc zmniejszenie TLCO jest zwykle proporcjonalne do zmniejszenia objętości płuc. Ponadto spadek TLCO koreluje z nasileniem ILD w HRCT i jest złym rokowniczo predyktorem.
- Test 6-minutowego chodu (6MWT – 6-minute walk test) – jest wystandaryzowany i dość łatwy do przeprowadzenia: podczas marszu trwającego 6 minut ocenia się przebyty dystans (w metrach) oraz stopień obniżenia wysycenia krwi tętniczej tlenem mierzony pulsoksymetrem (delta SaO2). Ocena SaO2 może być problematyczna u pacjentów z SSc ze względu na objaw Raynauda i słabą perfuzję palca. Jeśli dostępna jest odpowiednia nasadka, skuteczniejszy może być pomiar za pomocą klipsa przymocowanego do płatka ucha. Desaturacja podczas wysiłku jest wspólna zarówno dla ILD, jak i nadciśnienia płucnego.
Możliwości terapeutyczne w SSc
Relatywnie gorsze w porównaniu z innymi układowymi chorobami tkanki łącznej wyniki terapii SSc wiążą się z brakiem leczenia przyczynowego. Mimo znacznego postępu badań wciąż nie opracowano skutecznego leczenia przeciwzwłóknieniowego, a terapia jest uzależniona od stopnia zajęcia poszczególnych narządów i układów (tzw. terapia narządowo swoista). Szczególnie ważne jest leczenie farmakologiczne chorób układu oddechowego (śródmiąższowe włóknienie płuc) oraz sercowo-naczyniowego (tętnicze nadciśnienie płucne, zaburzenia rytmu serca i przewodzenia) ze względu na bezpośredni wpływ na długość życia pacjentów. Z drugiej strony zajęcie skóry (postępujące stwardnienie, objaw Raynauda i owrzodzenia) oraz układu ruchu (zapalenia stawów i ścięgien) mogą istotnie ograniczać jakość życia pacjentów, zwłaszcza na początku choroby. Stopień i dynamika zmian skórnych stanowią ponadto wykładnik aktywności SSc oraz korelują z zajęciem narządów wewnętrznych i śmiertelnością, przede wszystkim w postaci uogólnionej choroby.
W monitorowaniu postępu zmian skórnych stosuje się powszechnie tzw. zmodyfikowaną skalę Rodnana (mRSS – modified Rodnan Skin Score) 14 . Przeprowadzając badanie z wykorzystaniem mRSS, ocenia się napięcie skóry (0 – prawidłowe, 1 – łagodne, 2 – umiarkowane, 3 – znacznie zwiększone) w 17 obszarach obejmujących całą powierzchnię ciała. Wynik skali mieści się w zakresie od 0 do 51 pkt, przy czym wartości powyżej 20 pkt świadczą o znacznym zaawansowaniu choroby. W ostatnim czasie (pandemia COVID-19) wykazano przydatność uproszczonego testu, który pacjent może wykonać samodzielnie (PASTUL – Patient self-Assessment of Skin Thickness in Upper Limb). Ocenia się w nim jedynie 4 obszary na każdej kończynie górnej (palce, ręka, przedramię i ramię; zakres wartości 0-24 pkt). PASTUL może stanowić cenne narzędzie diagnostyczno-monitorujące w czasie teleporady 15 .
Leki stosowane w terapii SSc wybranych układów i narządów zestawiono w tabeli 4.
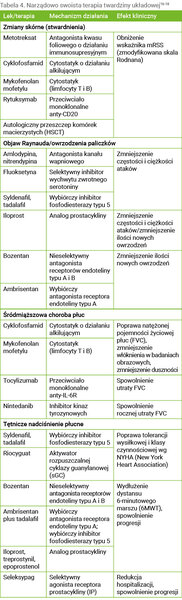
Tabela 4. Narządowo swoista terapia twardziny układowej16-18
W ostatnim czasie duże nadzieje wiąże się z autologicznym przeszczepieniem komórek macierzystych (HSCT – hematopoietic stem cell transplantation). Według europejskiego rejestru autologicznych przeszczepień szpiku (EBMT – European Society for Blood and Marrow Transplantation) w ostatnich latach w Europie wykonano łącznie 2809 procedur – najwięcej u pacjentów ze stwardnieniem rozsianym (50%), na drugim miejscu znalazły się osoby z chorobami tkanki łącznej (27%), wśród których znaczącą większość stanowią chorzy na SSc (79%) 19 .
W ciągu ostatnich 20 lat terapia HSCT była adresowana do pacjentów z ciężką i szybko postępującą SSc 20 . Według danych z międzynarodowych rejestrów liczba pacjentów po przeszczepie z roku na rok stopniowo wzrasta, co jest również dowodem na dobre wyniki związane z zastosowaniem tej opcji terapeutycznej 21 .
Wskazania do HSCT obejmują pacjentów 20, 22, 23 :
- z potwierdzoną diagnozą SSc, zgodnie z kryteriami ACR/EULAR z 2013 r. 9
- z układową postacią choroby, którzy w ocenie mRSS uzyskali co najmniej 16 pkt oraz doświadczyli pogorszenia o co najmniej 25% w ciągu ostatnich 6 miesięcy w trakcie leczenia immunosupresyjnego
- ze śródmiąższowym zajęciem płuc, ze spadkiem FVC lub TLCO o więcej niż 10% w ostatnich 6 miesiącach, w trakcie leczenia immunosupresyjnego.
Główne przeciwwskazania do HSCT obejmują 20, 22, 23 : podeszły wiek, ciężkie zaburzenia psychiczne, infekcje, choroby nowotworowe, niewydolność wątroby, nerek, układu oddechowego lub znaczne uszkodzenie serca.
O skuteczności HSCT u chorych na SSc mogą świadczyć wyniki badań ASTIS (Autologous Stem Cell Transplantation International Scleroderma) i SCOT (Scleroderma: Cyclophosphamide Or Transplantation), w których poddano leczeniu 231 pacjentów, z czego połowa miała wykonane HSCT, a druga połowa była leczona cyklofosfamidem. Odsetek przeżyć wyniósł 80% dla HSCT i 65% dla cyklofosfamidu w obserwacji 4-letniej 22 . W innej kohorcie wynik różnił się jeszcze istotniej i wyniósł 86% dla HSCT i 51% dla cyklofosfamidu w obserwacji 6-letniej 24 .
Doświadczenia z allogenicznym przeszczepieniem szpiku ograniczają się do pojedynczych opisów przypadków, dlatego dowody kliniczne są niewystarczające, aby zalecać ten sposób transplantacji szpiku u chorych na SSc 21, 25, 26 .
Zasady postępowania w SSc-ILD
Zasadniczym postępowaniem w SSc-ILD jest leczenie immunosupresyjne. W randomizowanych badaniach klinicznych nad SSc-ILD wykazano, że u chorych leczonych cyklofosfamidem uzyskano wolniejszy spadek FVC po roku leczenia w porównaniu z grupą placebo, a w badaniu oceniającym skuteczność 2-letniego leczenia mykofenolanem mofetylu w odniesieniu do rocznego leczenia cyklofosfamidem wykazano porównywalny efekt działania obu leków przy mniejszej toksyczności pierwszego z nich 17 .
Interesującą nową opcją terapeutyczną w SSc-ILD jest zastosowanie tocylizumabu – humanizowanego przeciwciała monoklonalnego wiążącego receptory dla interleukiny 6, leku biologicznego zarejestrowanego wcześniej w reumatoidalnym zapaleniu stawów (RZS), młodzieńczym idiopatycznym zapaleniu stawów, olbrzymiokomórkowym zapaleniu tętnic, a ostatnio również w najcięższych postaciach COVID-19. Tocylizumab okazał się nieskuteczny w hamowaniu progresji zmian skórnych, jednak jego podanie wiązało się z istotną statystycznie redukcją pogorszenia czynności płuc (hamowanie spadku FVC). Uzyskane dane pozwoliły na rejestrację przez amerykańską Agencję ds. Żywności i Leków (FDA – Food and Drug Administration) w 2021 r. tocylizumabu w postaci podskórnej u pacjentów z SSc-ILD 27 .
Kolejnym obiecującym lekiem biologicznym stosowanym dotychczas u pacjentów z RZS, który potencjalnie może okazać się skuteczny w terapii ILD związanej z SSc, jest rytuksymab – przeciwciało monoklonalne powodujące deplecję limfocytów B poprzez wiązanie się z przezbłonowym antygenem CD20. W niewielkim japońskim badaniu klinicznym rytuksymab poprawiał zarówno stan skóry (obniżenie wskaźnika mRSS), jak i wyniki badań czynnościowych (FVC) i obraz radiologiczny płuc 28 .
Obserwacje pacjentów z SSc-ILD wskazują, że ok. 30% chorych doświadcza progresji ILD w pierwszych 12 miesiącach obserwacji, a nawet 67% w okresie średnio 5 lat 29 . Pacjent z rozpoznaniem SSc-ILD powinien być monitorowany. Monitorowanie obejmuje ocenę objawów (tolerancji wysiłku, nasilenia kaszlu), szeroko pojęte badania czynnościowe płuc (np. FVC, TLCO, 6MWT) oraz w uzasadnionych przypadkach HRCT klatki piersiowej. Nie ma ustalonych wytycznych dotyczących częstości wykonywania badań kontrolnych w ILD. Zasadne wydaje się planowanie oceny klinicznej i czynnościowej (spirometria, dyfuzja) co 6-12 miesięcy, natomiast radiologicznej (HRCT) co rok-2 lata. W wybranych przypadkach, gdy narastają objawy lub istnieją czynniki ryzyka progresji, monitorowanie należy zaplanować indywidualnie.
Chorzy, u których stwierdza się progresję śródmiąższowego włóknienia płuc, powinni być kwalifikowani do leczenia antyfibrotycznego nintedanibem (patrz niżej).
W wybranych przypadkach, gdy ILD postępuje, a pacjent nie ma przeciwwskazań, należy rozważyć przeszczepienie płuc. Powinno się je również wziąć pod uwagę u dorosłych z zaawansowaną chorobą płuc, u których istnieje wysokie ryzyko śmierci w ciągu kolejnych 2 lat (przy jednoczesnym wysokim prawdopodobieństwie przeżycia krótkoterminowego i długoterminowego po wykonaniu zabiegu).
W codziennej praktyce przeszczepienie płuc rzadko wykonuje się u chorych na SSc. W międzynarodowym rejestrze w 2016 r. pacjenci z tą chorobą stanowili tylko 1,1% wśród wszystkich chorych poddanych tej procedurze 30 . Przeżycie po przeszczepieniu płuc jest niższe niż po transplantacjach innych narządów. Według OPTN (Organ Procurement and Transplantation Network) 5 lat przeżywa 47% chorych w porównaniu z 80% po przeszczepieniu nerki lub 72% po transplantacji serca lub wątroby.
SSc jako choroba ogólnoustrojowa prowadząca do dysfunkcji wielu narządów stanowi duże wyzwanie dla zespołów transplantacyjnych. Pacjent przed przeszczepieniem wymaga starannego przygotowania. W rezultacie wiele ośrodków jest niechętnych, aby przeprowadzać transplantacje płuc w tej grupie chorych.
Większość pacjentów z SSc kierowanych do przeszczepu cierpi na nadciśnienie płucne, 1/3 chorych ma tylko zmiany śródmiąższowe w płucach. Wskaźniki przeżycia po roku, 3 i 5 latach od transplantacji wynoszą odpowiednio 81%, 68% i 61%. Czynnikami wpływającymi na gorsze przeżycie po przeszczepieniu płuc są płeć żeńska oraz stwierdzenie nadciśnienia płucnego (gdy występowały oba te czynniki, ryzyko śmierci było 3-krotnie większe niż u mężczyzn bez PAH). Nowe doniesienia pokazują jednak, że wskaźniki przeżycia po przeszczepieniu płuc i okres wolny od przewlekłej dysfunkcji graftu u chorych z SSc były podobne jak w przypadku pacjentów kwalifikowanych do transplantacji w związku z innymi chorobami płuc 31 . Niemniej jest to wciąż grupa o wysokim ryzyku niepowodzenia, co znajduje odzwierciedlenie również w polskich statystykach. W ośrodku w Zabrzu w latach 2004-2018 przeszczepiono płuca u 47 chorych z ILD, przy czym żaden z nich nie chorował na SSc 32 . Ośrodek w Gdańsku w ostatnich latach wykonał przeszczepienie płuc u 1 osoby z SSc, natomiast w ośrodku w Szczecinie nie było takiego przypadku.
Koncepcja postępującego włóknienia płuc
Mimo optymalnego leczenia choroby podstawowej u ok. 40% chorych z SSc-ILD śródmiąższowe włóknienie płuc postępuje. Patomechanizm postępującego włóknienia płuc w twardzinie jest porównywalny do tego w przebiegu idiopatycznego włóknienia płuc (IPF – idiopathic pulmonary fibrosis), a rokowanie jest podobnie złe. Od czasu publikacji wyników badań SENSCIS (Safety and Efficacy of Nintedanib in Systemic Sclerosis) i INBUILD (Efficacy and Safety of Nintedanib in Patients With Progressive Fibrosing Interstitial Lung Disease) u takich chorych rozpoznaje się fenotyp postępującego włóknienia w przebiegu SSc-ILD (PPF – progressive pulmonary fibrosis) 33 . Wyniki wymienionych badań pokazały, że niezależnie od rozpoznania choroby podstawowej, w tym SSc, w grupie chorych, którzy spełniają kryteria rozpoznania fenotypu postępującego włóknienia płuc, zastosowanie nintedanibu (doustnego inhibitora kinazy tyrozynowej o działaniu antyfibrotycznym) zmniejsza tempo progresji ILD mierzonej spadkiem FVC 34, 35 .
Aby rozpoznać PPF, należy wykazać występowanie 2 z 3 cech:
- narastanie objawów ze strony układu oddechowego
- pogarszanie się parametrów czynnościowych układu oddechowego (największą rolę odgrywa pogarszanie się FVC)
- narastanie cech śródmiąższowego włóknienia w obrazie HRCT klatki piersiowej.
Opieka nad chorymi z postępującym włóknieniem płuc stanowi wyzwanie, które wymaga interdyscyplinarnego podejścia (zwłaszcza współpracy pulmonologów i reumatologów) oraz odmiennego leczenia – opartego na leku antyfibrotycznym.
W badaniu SENSCIS wykazano również, że skojarzenie leku antyfibrotycznego (nintedanibu) z lekiem immunosupresyjnym jest bezpieczne i może przynieść korzystne efekty w zakresie spowolnienia tempa progresji śródmiąższowego włóknienia płuc. Do badania włączano chorych na SSc-ILD, otrzymujących w ciągu ostatnich 6 miesięcy stabilną dawkę mykofenolanu mofetylu, metotreksatu lub ≤10 mg prednizonu. Ostatecznie mniej więcej połowa chorych była leczona pierwszym z wymienionych leków. Zaobserwowano, że w grupie placebo spadek FVC był mniejszy u pacjentów otrzymujących mykofenolan mofetylu, co sugeruje potencjalny korzystny wpływ tego leku. Ponadto pacjenci leczeni zarówno nintedanibem, jak i mykofenolanem mofetylu charakteryzowali się najwolniejszym tempem spadku FVC, co wskazuje na potencjalną korzyść z leczenia skojarzonego SSc-ILD.
Jeśli pomimo leczenia stwierdza się cechy postępującego włóknienia płuc w przebiegu SSc-ILD, należy rozważyć leczenie antyfibrotyczne nintedanibem, który w kwietniu 2020 r. został zatwierdzony jako pierwszy (i obecnie jedyny) lek spowalniający tempo pogarszania się czynności płuc u dorosłych pacjentów z postępującym włóknieniem w przebiegu SSc-ILD. Terapia ta od 1 lipca 2022 r. jest refundowana dla polskich pacjentów w ramach programu lekowego NFZ (program lekowy B.135).
Postuluje się, że pirfenidon – lek antyfibrotyczny, który jako pierwszy został zarejestrowany do leczenia chorych na IPF – może mieć podobny do nintedanibu potencjał w spowalnianiu włóknienia w innych niż IPF śródmiąższowych chorobach płuc przebiegających z włóknieniem, w tym w SSc-ILD. Jednak obecnie pirfenidon ma jedynie rejestrację do leczenia IPF. Oczekiwane są publikacje wyników badań klinicznych z zastosowaniem tego leku u chorych z SSc-ILD.
Istnieje potrzeba dalszego poszukiwania skutecznych leków w terapii SSc-ILD, zarówno o przebiegu NSIP, jak i postępującego włóknienia.
Podsumowanie
SSc jest układową chorobą tkanki łącznej, w której oprócz typowego zajęcia skóry i układu ruchu bardzo często (nawet w 90% przypadków) dochodzi do uszkodzenia płuc, przebiegającego najczęściej w postaci ILD. Podobnie jak we wszystkich chorobach przewlekłych, wpływających na długość życia pacjenta, podstawowe znaczenie rokownicze ma jak najwcześniejsze rozpoznanie. W przypadku SSc dotyczy to zarówno wczesnej diagnozy samej twardziny (objaw Raynauda, obrzęk palców rąk), jak i pojawiającego się z upływem czasu zajęcia płuc. Ponieważ objawy kliniczne ze strony układu oddechowego (pogorszenie tolerancji wysiłku, duszność, kaszel) występują późno, od samego początku istotne jest zwrócenie szczególnej uwagi na pacjentów obciążonych największym ryzykiem włóknienia płuc – tych z uogólnioną postacią twardziny oraz obecnością specyficznych przeciwciał przeciwjądrowych (Scl-70, Th/To, U12/U12-RNP). U chorych z tej grupy należy rozważyć konsultację pulmonologiczną, wykonanie HRCT klatki piersiowej (podstawa rozpoznania SSc-ILD) oraz testów czynnościowych płuc pomimo braku klinicznych objawów zajęcia układu oddechowego. Trwają również poszukiwania coraz bardziej specyficznych biomarkerów wczesnej twardziny oraz przedklinicznych etapów śródmiąższowej choroby płuc.
Ze względu na nie do końca poznaną patogenezę SSc, nie dysponujemy nadal efektywną terapią przyczynową. Leczenie SSc staje się jednak coraz skuteczniejsze (poprawa jakości i długości życia), co jest związane z uzależnieniem stosowanej terapii od zajęcia poszczególnych narządów i układów (terapia narządowo swoista), rejestracji nowych leków oraz doskonalenia procedur terapeutycznych (HSCT, przeszczepienie płuc). Dotyczy to w szczególności najcięższych postaci SSc powikłanych śródmiąższowym włóknieniem płuc. Oprócz stosowanych dotychczas rutynowo leków o działaniu cytostatycznym (cyklofosfamid, mykofenolan mofetylu) w ostatnich latach zostały zarejestrowane nowe leki, w przypadku których udowodniono spowolnienie progresji SSc-ILD (nintedanib, tocylizumab). Warto również podkreślić, że te najnowocześniejsze leki, wysoko wybiórczo ingerujące w działanie układu immunologicznego, stają się coraz bardziej dostępne dla polskich pacjentów z SSc oraz zależnym od twardziny śródmiąższowym włóknieniem płuc (program lekowy NFZ dla nintedanibu).
Abstract
Systemic sclerosis and pulmonary problems: recommendations not only for rheumatologists
Systemic sclerosis (SSc) is a complex, multi-system, autoimmune connective tissue disease. Even though the exact etiology of SSc is unclear, it has been established that microvascular (endothelial) injuries, formation of specific autoantibodies and organ fibrosis play a crucial role in the development of the disease. SSc is classified into two main subtypes: limited cutaneous systemic sclerosis (lSSc) and diffuse cutaneous systemic sclerosis (dcSSc). Skin thickening is the most common clinical symptom of SSc and it is considered to be the most important prognostic factor and marker of the disease activity. The 2013 ACR/EULAR classification criteria help in the identification of early SSc, thus making it possible to initiate treatment at an earlier stage of the disease. Interstitial lung disease (ILD) and cardiac involvement are currently recognized as the leading causes of SSc-associated mortality. The early detection of ILD and an aggressive therapeutic intervention could help to improve patient outcomes. High-resolution computed tomography is the most effective method of diagnosing ILD in SSc patients and an important tool for monitoring the disease progression. Treatment for SSc should be implemented to target organ-specific complications of the disease. Cyclophosphamide and mycophenolate mofetil are the most commonly prescribed immunosuppressive treatments for SSc-ILD. Recently, nintedanib (an antifibrotic agent) and tocilizumab (an anti-IL6 receptor antibody, FDA only) have been approved for SSc-ILD patients. They are indicated for slowing the rate of decline in pulmonary function. Further research is required to identify the best therapeutic algorithms (including combined immunosuppressive and antifibrotic treatment) aimed at stabilizing or even improving pulmonary function in the course of SSc-ILD.
- 1. World Scleroderma Foundation [Internet]. What is Scleroderma? http://www.worldsclerofound.org/what-is-ss
- 2. Bergamasco A, Hartmann N, Wallace L, et al. Epidemiology of systemic sclerosis and systemic sclerosis-associated interstitial lung disease. Clin Epidemiol 2019;11:257-73
- 3. Sznyk A, Bukowski H, Skrzekowska-Baran I. Twardzina układowa ze szczególnym uwzględnieniem choroby śródmiąższowej płuc w Polsce. Epidemiologia w latach 2008-2018. Instytut Innowacji i Odpowiedzialnego Rozwoju INNOWO, 2020
- 4. Ezdrowie. Zestawienia [Internet]. Liczba pacjentów z rozpoznaniem twardziny układowej oraz innych chorób śródmiąższowych płuc w latach 2009-2020. https://ezdrowie.gov.pl/portal/home/badania-i-dane/zdrowe-dane/zestawienia/liczba-pacjentow-z-twardzina-ukladowa-oraz-innymi-chorobami-srodmiazszowymi-pluc
- 5. Steen VD, Medsger TA. Changes in causes of death in systemic sclerosis, 1972-2002. Ann Rheum Dis 2007;66(7):940-44
- 6. Davidsen JR, Miedema J, Wuyts W, et al. Economic Burden and Management of Systemic Sclerosis-Associated Interstitial Lung Disease in 8 European Countries: The BUILDup Delphi Consensus Study. Adv Ther 2021;38(1):521-40
- 7. Kucharz EJ, Kopeć-Mędrek M. Systemic sclerosis sine scleroderma. Adv Clin Exp Med 2017;26(5):875-80
- 8. Sierakowski S, Sierakowska M, Goncerz G. Interna. Mały Podręcznik [Internet]. Rozdział: Twardzina układowa. https://www.mp.pl/interna/chapter/B16.II.16.5
- 9. Hoogen F van den, Khanna D, Fransen J, et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League against Rheumatism collaborative initiative. Arthritis Rheum 2013;65(11):2737-47
- 10. Minier T, Guiducci S, Bellando-Randone S, et al. Preliminary analysis of the very early diagnosis of systemic sclerosis (VEDOSS) EUSTAR multicentre study: evidence for puffy fingers as a pivotal sign for suspicion of systemic sclerosis. Ann Rheum Dis 2014;73(12):2087-93
- 11. Sobolewski P, Maślińska M, Wieczorek M, et al. Systemic sclerosis – multidisciplinary disease: clinical features and treatment. Reumatologia 2019;57(4):221-33
- 12. McNearney TA, Reveille JD, Fischbach M, et al. Pulmonary involvement in systemic sclerosis: associations with genetic, serologic, sociodemographic, and behavioral factors. Arthritis Rheum 2007;57(2):318-26
- 13. Suliman YA, Dobrota R, Huscher D, et al. Brief Report: Pulmonary Function Tests: High Rate of False-Negative Results in the Early Detection and Screening of Scleroderma-Related Interstitial Lung Disease. Arthritis Rheumatol 2015;67(12):3256-61
- 14. Zalewska J, Barczyńska T, Węgierska M, et al. Zastosowanie zmodyfikowanej skali Rodnana (mRSS) w codziennej praktyce reumatologicznej. Forum Reumatol 2016;2(3):125-9
- 15. Spierings J, Ong V, Denton CP. PASTUL questionnaire: a tool for self-assessment of scleroderma skin during the COVID-19 pandemic. Ann Rheum Dis 2021;80(6):819-20
- 16. Bukiri H, Volkmann ER. Current advances in the treatment of systemic sclerosis. Curr Opin Pharmacol 2022;64:102211
- 17. Tashkin DP, Roth MD, Clements PJ, et al. Mycophenolate mofetil versus oral cyclophosphamide in scleroderma-related interstitial lung disease (SLS II): a randomised controlled, double-blind, parallel group trial. Lancet Respir Med 2016;4(9):708-19
- 18. McHugh J. Tocilizumab prevents ILD progression in early SSc. Nat Rev Rheumatol 2021;17(4):188
- 19. Oliveira MC, Elias JB, Moraes DA de, et al. A review of hematopoietic stem cell transplantation for autoimmune diseases: multiple sclerosis, systemic sclerosis and Crohn’s disease. Position paper of the Brazilian Society of Bone Marrow Transplantation. Hematol Transfus Cell Ther 2021;43(1):65-86
- 20. Snowden JA, Saccardi R, Allez M, et al. Haematopoietic SCT in severe autoimmune diseases: updated guidelines of the European Group for Blood and Marrow Transplantation. Bone Marrow Transplant 2012;47(6):770-90
- 21. Snowden JA, Badoglio M, Labopin M, et al. Evolution, trends, outcomes, and economics of hematopoietic stem cell transplantation in severe autoimmune diseases. Blood Adv 2017;1(27):2742-55
- 22. Laar JM van, Farge D, Sont JK, et al. Autologous hematopoietic stem cell transplantation vs intravenous pulse cyclophosphamide in diffuse cutaneous systemic sclerosis: a randomized clinical trial. JAMA 2014;311(24):2490-8
- 23. Burt RK, Oliveira MC, Shah SJ, et al. Cardiac involvement and treatment-related mortality after non-myeloablative haemopoietic stem-cell transplantation with unselected autologous peripheral blood for patients with systemic sclerosis: a retrospective analysis. Lancet 2013;381(9872):1116-24
- 24. Sullivan KM, Goldmuntz EA, Keyes-Elstein L, et al. Myeloablative autologous stem-cell transplantation for severe scleroderma. N Engl J Med 2018;378(1):35-47
- 25. Loh Y, Oyama Y, Statkute L, et al. Non-myeloablative allogeneic hematopoietic stem cell transplantation for severe systemic sclerosis: graft-versus-autoimmunity without graft-versus-host disease? Bone Marrow Transplant 2007;39(7):435-7
- 26. Shiratsuchi M, Motomura S, Abe Y, et al. Long-term follow-up after nonmyeloablative allogeneic hematopoietic stem cell transplantation for systemic sclerosis. Clin Rheumatol 2008;27(9):1207-9
- 27. Genentech, Inc. ACTEMRA® (tocilizumab) injection, for intravenous or subcutaneous use Initial U.S. Approval: 2010 [Internet]. U.S Food and Drug Administration. https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/125472s044lbl.pdf
- 28. Ebata S, Yoshizaki A, Oba K, et al. Safety and efficacy of rituximab in systemic sclerosis (DESIRES): a double-blind, investigator-initiated, randomised, placebo-controlled trial. Lancet Rheumatol 2021;3(7):e489-97
- 29. Hoffmann-Vold A-M, Allanore Y, Alves M, et al. Progressive interstitial lung disease in patients with systemic sclerosis-associated interstitial lung disease in the EUSTAR database. Ann Rheum Dis 2021;80(2):219-27
- 30. Yusen RD, Edwards LB, Dipchand AI, et al. The Registry of the International Society for Heart and Lung Transplantation: Thirty-third Adult Lung and Heart-Lung Transplant Report-2016; Focus Theme: Primary Diagnostic Indications for Transplant. J Heart Lung Transplant 2016;35(10):1170-84
- 31. Pradère P, Tudorache I, Magnusson J, et al. Lung transplantation for scleroderma lung disease: An international, multicenter, observational cohort study. J Heart Lung Transplant 2018;37(7):903-11
- 32. Stącel T, Nęcki M, Antończyk R, et al. Effectiveness of Lung Transplantation in Patients With Interstitial Lung Diseases. Transplant Proc 2020;52(7):2143-8
- 33. Raghu G, Remy-Jardin M, Richeldi L, et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med 2022;205(9):e18-47
- 34. Distler O, Highland KB, Gahlemann M, et al. Nintedanib for Systemic Sclerosis-Associated Interstitial Lung Disease. N Engl J Med 2019;380(26):2518-28
- 35. Flaherty KR, Wells AU, Cottin V, et al. Nintedanib in Progressive Fibrosing Interstitial Lung Diseases. N Engl J Med 2019;381(18):1718-27


 Dodaj do ulubionych
Dodaj do ulubionych
