



Co znajdziesz w artykule?
- Omówienie postępowania w idiopatycznym zapaleniu płuc (IPF) na podstawie wytycznych ERS i ATS oraz wskazanie różnic w stosunku do zaleceń Fleischner Society i PTChP
- Kryteria diagnostyczne IPF oraz badania, które są pomocne w ustaleniu rozpoznania (obrazowe, serologiczne, histopatologiczne)
- Metody leczenia IPF obejmujące rehabilitację oddechową, stosowanie leków przeciwzwłóknieniowych i przeszczepienie płuc
Spis treści
Idiopatyczne włóknienie płuc jako problem kliniczny
Idiopatyczne włóknienie płuc (IPF – idiopathic pulmonary fibrosis), zwane też samoistnym włóknieniem płuc, stanowi rzadką chorobę układu oddechowego. W Europie zapadalność na nią szacowana jest na 2,3-7,4/100 000 rocznie. Należy do najczęściej rozpoznawanych śródmiąższowych chorób płuc z grupy śródmiąższowych samoistnych zapaleń płuc (około 50% wszystkich rozpoznań z tej grupy). Stanowi specyficzne zapalenie śródmiąższowe ograniczone do
tkanki płuc, o obrazie radiologicznym lub histopatologicznym tzw. zwykłego śródmiąższowego zapalenia płuc (UIP – usual interstitial pneumonia). Proces przebudowy miąższu płucnego w przebiegu IPF ma charakter postępujący.
Sam wzorzec UIP może występować w wielu innych jednostkach chorobowych, w tym zajmujących głównie układ oddechowy (np. zapalenie płuc z nadwrażliwości lub pylica azbestowa), jak również w chorobach układowych, w których zajęcie układu oddechowego jest jednym z elementów obrazu chorobowego (np. reumatoidalne zapalenie stawów [RZS] i zapalenie skórno-mięśniowe). Wreszcie UIP może być skutkiem uszkodzenia płuc w przebiegu niepożądanej reakcji polekowej.
Potencjalne liczne etiologie wzorca UIP stanowią o złożoności procesu diagnostycznego – w praktyce rozpoznanie jest diagnozą z wykluczenia innych znanych przyczyn włóknienia. Poprawnie przeprowadzona diagnostyka różnicowa pozwala również na ewentualne wprowadzenie terapii celowanej w przyczynę wtórnego włóknienia płuc (np. eliminacja antygenu w zapaleniach płuc z nadwrażliwości), a nie tylko na leczenie celowane w spowolnienie procesu włóknienia płuc.
W pracy zostanie omówione postępowanie diagnostyczne i lecznicze w idiopatycznym włóknieniu płuc.
Diagnostyka
Obraz kliniczny
Idiopatyczne włóknienie płuc jest chorobą osób w dojrzałym wieku – praktycznie nie występuje przed 50 r.ż., a większość przypadków jest rozpoznawana u osób w 7 dekadzie życia. Reguła ta nie dotyczy rodzinnej postaci choroby, która może wystąpić we wcześniejszym wieku. Ponadto w tej postaci w każdym kolejnym pokoleniu choroba ujawnia się wcześniej i może mieć szybszy przebieg 1, 2 .
Większość chorych na IPF jest płci męskiej. W wywiadzie zwraca również uwagę narażenie na dym tytoniowy – ryzyko rozwoju choroby, podobnie jak w raku płuca, wzrasta przy przekroczeniu 20 paczkolat 1, 2 .
Pacjenci zgłaszają duszność, ograniczenie tolerancji wysiłku oraz suchy kaszel, które nasilają się wraz z czasem trwania choroby. U niektórych występują ponadto ogólnoustrojowe objawy choroby, np. utrata masy ciała czy osłabienie. Stwierdzenie innych objawów ogólnoustrojowych, takich jak bóle kostno-stawowe, osłabienie siły mięśniowej, stany podgorączkowe, zmiany skórne i śluzówkowe, nakazuje zastanowić się nad potencjalnie wtórnym charakterem zmian płucnych 1, 3 .
W badaniu przedmiotowym u pacjentów z IPF najczęściej stwierdza się symetryczne trzeszczenia, głównie wdechowe, u podstawy obu płuc. Lokalizacja może się zmieniać, tj. trzeszczenia mogą się pojawiać w wyższych partiach płuc wraz z postępem procesu chorobowego. Drugim często stwierdzanym odchyleniem jest przyspieszenie i spłycenie oddechu. W zaawansowanych postaciach choroby można zaobserwować palce pałeczkowate. Wraz z rozwojem powikłań choroby pojawiają się charakterystyczne dla nich objawy, np. sinica w niewydolności oddechowej lub objawy niewydolności serca w przebiegu nadciśnienia płucnego 1, 2 .
Kryteria diagnostyczne
Przed 2000 r. nie istniały jednolite i powszechnie uznawane kryteria rozpoznania IPF. Pierwsze uniwersalne kryteria powstały na drodze konsensusu European Respiratory Society (ERS) oraz American Thoracic Society (ATS). Wraz z postępem wiedzy podlegały one ewolucji, w związku z czym opublikowano dwie aktualizacje – w 2011 i 2018 r. 2, 4 Ostatni konsensus po aktualizacjach, przyjęty nie tylko przez ERS i ATS, lecz także przez tożsame towarzystwa naukowe japońskie i Ameryki Łacińskiej, stanowi najszerzej uznawane kryteria diagnostyczne choroby. Same kryteria diagnostyczne były również tematem publikacji ekspertów Fleischner Society 3 .
Dodatkowo Polskie Towarzystwo Chorób Płuc (PTChP) opublikowało w 2020 r. własną modyfikację wytycznych na podstawie nowych dowodów naukowych 1 . W artykule oprzemy się głównie na światowych kryteriach z 2018 r., zaznaczymy jednak różnice w sytuacjach, w których wytyczne Fleischner Society oraz PTChP rekomendują inne postępowanie.
Konsensus z 2018 r. wyróżnia 3 kryteria diagnostyczne dla IPF, przy czym dla rozpoznania niezbędne jest spełnienie kryterium 1 i dowolnego z dwóch pozostałych:
1. Wykluczenie innych potencjalnych przyczyn śródmiąższowych chorób płuc
2. Obecność wzorca UIP w tomografii komputerowej wysokiej rozdzielczości (HRCT – high resolution computed tomography)
3. Adekwatne zestawienie obrazu HRCT oraz histologicznej oceny bioptatu płuca (tab. 1).
W kolejności omówione zostaną wyżej wymienione kryteria.
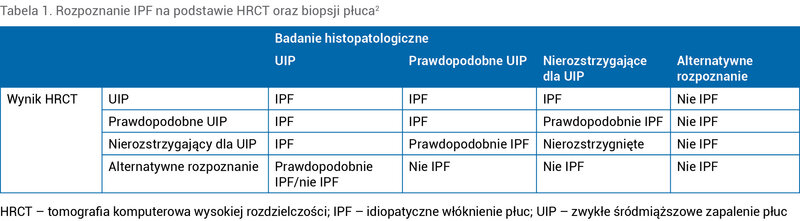
Tabela 1. Rozpoznanie IPF na podstawie HRCT oraz biopsji płuca2
Eksperci ERS/ATS w celu wykluczenia przyczyn wtórnego UIP zalecają zebranie szczegółowego wywiadu na temat leków przyjmowanych aktualnie oraz w przeszłości. W kwestii substancji chemicznych mogących powodować wystąpienie objawów UIP można się posiłkować danymi dostępnymi w witrynie Pneumotox online (www.pneumotox.com).
Ponadto zaleca się zebranie wywiadu dotyczącego potencjalnych ekspozycji na substancje środowiskowe występujące w miejscu zamieszkania, pracy oraz miejscach, które pacjent często odwiedza. W tym celu można zastosować usystematyzowane kwestionariusze ukierunkowane na narażenia, które mogą wywołać pylicę, jak również zapalenie płuc z nadwrażliwości. Szczegółowa konstrukcja kwestionariuszy zależy od lokalnych uwarunkowań kulturowych oraz geograficznych.
Odrębną kwestię stanowi diagnostyka serologiczna. Autorzy konsensusu ERS/ATS sugerują wykonanie badań w kierunku chorób tkanki łącznej niezależnie od ewentualnych objawów klinicznych tych schorzeń. Wynika to z możliwości wyprzedzania przez zmiany płucne manifestacji ogólnoustrojowej. Najczęściej wymieniane są badania w kierunku przeciwciał przeciwjądrowych (ANA – antinuclear antibodies), czynnika reumatoidalnego (RF – rheumatoid factor), przeciwciał przeciwko cyklicznym cytrulinowanym peptydom (anty-CCP – anti-cyclic citrullinated peptide antibodies), ewentualnie panel przeciwciał specyficznych dla zapaleń mięśni. Dodatkowo w przypadku podejrzenia zapaleń mięśni można się wspierać oznaczeniem stężenia mioglobiny oraz aktywności aldolazy i kinazy fosfokreatynowej. Polskie wytyczne wymieniają ponadto przeciwciała przeciw cytoplazmie neutrofilów (ANCA – anti-neutrophil cytoplasmic antibodies) w zakresie panelu badań w kierunku wtórnych przyczyn włóknienia płuc 2 .
W przypadku różnicowania z zapaleniem płuc z nadwrażliwości warto wykonać panel przeciwciał w tym kierunku, tzw. precypityn, jednak dostępność tego badania jest ograniczona.
Eksperci Fleischner Society w diagnostyce różnicowej zwracają uwagę na istnienie śródmiąższowego zapalenia płuc z cechami autoimmunizacji (IPAF – interstitial pneumonia with autoimmune features), tj. jednostki chorobowej, w której do obrazu śródmiąższowego włóknienia płuc dołączają się pewne cechy chorób tkanki łącznej, ale nie są spełnione wszystkie kryteria potrzebne do ich rozpoznania. U pacjentów z takimi cechami klinicznymi sugeruje się niewykluczanie IPF z uwagi na brak jednoznacznych kryteriów diagnostycznych dla IPAF. Towarzystwo uznaje za niepewną rolę precypityn w diagnostyce różnicowej z zapaleniem płuc z nadwrażliwości 3 .
Rekomendacje PTChP również nie przewidują konieczności wykonania badania swoistych precypityn u wszystkich osób z podejrzeniem IPF. Inne wysoce specjalistyczne oznaczenia o potencjalnym zastosowaniu w diagnostyce różnicowej chorób śródmiąższowych (np. metaloproteinaza 7 [MMP7 – matrix metalloproteinase 7], białko surfaktantu D [SPD – surfactant protein D], ligand 18 chemokin [CCL18 – CC-chemokine ligand 18], białko Krebs von den Lungen 6 [KL6]) nie powinny być stosowane z uwagi na duży odsetek fałszywie pozytywnych i fałszywie negatywnych wyników 1 .
Podsumowanie dotyczące badań serologicznych w diagnostyce IPF przedstawiono w tabeli 2.

Tabela 2. Badania serologiczne w diagnostyce IPF
Podstawowe badanie obrazowe w diagnostyce IPF stanowi HRCT. Kryteria z 2018 r. wyróżniają 4 kategorie oceny HRCT w kierunku IPF: obraz UIP, prawdopodobnego UIP, nierozstrzygający dla UIP, sugerujący alternatywne rozpoznanie. W kolejności kategorie te scharakteryzowane są następująco:
1. Obraz UIP:
- lokalizacja zmian przypodstawna oraz podopłucnowa
- heterogenność dystrybucji zmian
- obecność plastra miodu (honeycombing) z towarzyszącymi rozstrzeniami z pociągania oskrzeli lub oskrzelików (traction bronchiectasis/bronchiolectasis) albo bez nich
2. Obraz prawdopodobnego UIP:
- lokalizacja zmian przypodstawna oraz podopłucnowa
- zmiany siateczkowate z towarzyszącymi rozstrzeniami z pociągania oskrzeli lub oskrzelików
- możliwe nienasilone cechy mlecznej szyby
3. Obraz nierozstrzygający dla UIP:
- lokalizacja zmian przypodstawna oraz podopłucnowa
- delikatna siateczka, możliwa mleczna szyba (określane jako wczesne UIP)
- cechy tomografii komputerowej (TK) lub dystrybucja zmian włóknistych niesugerująca żadnego rozpoznania
4. Obraz sugerujący alternatywne rozpoznanie; na kategorię tę składają się 3 elementy:
- zmiany sugerujące inne rozpoznanie
- torbiele
- mozaikowate upowietrznienie
- przewaga mlecznej szyby
- zmiany drobnoguzkowe
- guzki środka zrazika
- guzki
- konsolidacje
- dominująca lokalizacja
- około pęczków naczyniowo-oskrzelowych
- około naczyń limfatycznych
- w polach płucnych górnych i środkowych
- inne
- płytki opłucnowe
- poszerzony przełyk
- obecność zaburzenia struktury kostnej obojczyków
- obecność wysięku w opłucnej
- znaczne powiększenie węzłów chłonnych.
Jedną z podstawowych cech radiologicznych UIP jest obraz plastra miodu. Warte podkreślenia są trudności z jego prawidłowym rozpoznaniem, zwłaszcza w kontekście współistnienia takich zmian, jak rozstrzenie z pociągania i rozedma. Eksperci Fleischner Society za wystarczające do rozpoznania plastra miodu uznają pojedynczą podopłucnową warstwę 2 lub 3 sąsiadujących ze sobą torbieli 3 .
Szczególnie rozbudowana ostatnia kategoria naprowadza na możliwe inne przyczyny dolegliwości pacjenta, np. twardzinę układową (zmiany w przełyku), RZS (zmiany kostne), histiocytozę lub limfangioleiomiomatozę (zmiany torbielowate). Wśród chorób tkanki łącznej, których objawy występują w układzie oddechowym, główne znaczenie w różnicowaniu ma reumatoidalne zapalenie stawów z uwagi na duże rozpowszechnienie w populacji. W przeciwieństwie do innych chorób z tej grupy jego najczęstszą manifestacją jest właśnie UIP, a nie niespecyficzne zapalenie śródmiąższowe płuc (NSIP – nonspecific interstitial pneumonia). Ponadto zmiany płucne w RZS, jak wspomniano już wyżej, mogą wyprzedzać w czasie pojawienie się charakterystycznych objawów stawowych.
U pacjentów poddanych biopsji płuca analogicznie do oceny obrazu HRCT wprowadzono 4 kategorie oceny histopatologicznej pobranego materiału: UIP, prawdopodobne UIP, nierozstrzygające dla UIP, alternatywne rozpoznanie. Przedstawiają się one następująco:
1. UIP:
- włóknienie z zaburzeniem architektoniki
- lokalizacja podopłucnowa i okołoprzegrodowa
- rozproszone ogniska włóknienia w miąższu płuca
- ogniska fibroblastów
- brak cech sugerujących inne rozpoznanie
2. Prawdopodobne UIP:
- niektóre cechy z kategorii UIP, ale niewystarczające dla rozpoznania UIP, w tym niestwierdzenie cech sugerujących inne rozpoznanie albo
- obecność tylko plastra miodu
3. Nierozstrzygające dla UIP:
- zwłóknienie z zaburzeniami architektoniki lub bez nich sugerujące UIP, jak również zmiany inne niż UIP
- cechy UIP i cechy sugerujące alternatywne rozpoznanie
4. Alternatywne rozpoznanie:
- cechy innych śródmiąższowych zapaleń płuc
- cechy histologiczne innych chorób śródmiąższowych.
Biopsja płuca oraz płukanie oskrzelowo-pęcherzykowe
Wytyczne ERS/ATS poruszają kwestię diagnostyki inwazyjnej, tj. biopsji płuca chirurgicznej, biopsji przezoskrzelowej, kriobiopsji oraz bronchoskopii z wykonaniem płukania oskrzelowo-pęcherzykowego (BAL – bronchoalveolar lavage). Ewentualna kwalifikacja do tych procedur diagnostycznych zależy od obrazu HRCT. Uzyskanie pewnego obrazu UIP zwalnia od wykonania biopsji chirurgicznej, biopsji przezoskrzelowej, kriobiopsji i bronchoskopii z pobraniem BAL. W przypadku obrazu prawdopodobnego, nierozstrzygającego lub sugerującego alternatywne rozpoznanie biopsja chirurgiczna i BAL stanowią sugerowane postępowanie. Biopsja przezoskrzelowa oraz kriobiopsja w tym przypadku nie mają rekomendacji za ani przeciw. Wynika to z możliwości wywołania nagłego zaostrzenia IPF w wyniku diagnostyki inwazyjnej. Powikłanie to obarczone jest wysoką śmiertelnością 2 .
Stanowisko PTChP w kwestii diagnostyki inwazyjnej częściowo się różni. W polskich wytycznych sugeruje się, by również obraz prawdopodobnego UIP w HRCT, przy braku zmian wskazujących na alternatywne rozpoznanie, wystarczał do rozpoznania IPF bez konieczności wykonywania biopsji płuca. Ponadto eksperci PTChP wskazują, by materiał uzyskany drogą kriobiopsji traktować na równi z uzyskanym za pomocą biopsji chirurgicznej 1 .
Rekomendacje PTChP dotyczą również w szerszym stopniu kwalifikacji do diagnostyki inwazyjnej, rozumianej tutaj jako biopsja płuca. Czynnikami ryzyka powikłań diagnostyki zabiegowej są niskie wartości współczynnika transferu płucnego dla tlenku węgla (TLCO – lung transfer factor for carbon monoxide), natężona pojemność życiowa (FVC – forced vital capacity) <50% i ciężka hipoksemia. Z tego powodu rekomendowane jest wykonanie gazometrii krwi tętniczej, spirometrii oraz oceny TLCO w ramach kwalifikacji do zabiegu 1 .
Rola zespołu wielospecjalistycznego
W wielu wytycznych, jak również publikacjach dotyczących IPF postuluje się główną rolę zespołu wielospecjalistycznego w diagnostyce IPF. Rola ta rośnie zwłaszcza w przypadku, gdy nie ma możliwości rozpoznania pewnego UIP w obrazie radiologicznym. W podstawowym składzie zespołu powinni się znaleźć pulmonolog oraz radiolog z doświadczeniem w diagnostyce chorób śródmiąższowych płuc, a także – w przypadku wykonywania biopsji płuc – patomorfolog. Dodatkowo w zależności od kontekstu klinicznego można skorzystać z doświadczenia innych specjalistów, wśród których wymienia się reumatologów, specjalistów medycyny pracy i kardiologów 1, 2 .
Konsultację reumatologiczną należy zaproponować zwłaszcza pacjentom z dodatnimi wynikami oznaczeń serologicznych w kierunku chorób tkanki łącznej, obrazem klinicznym sugerującym chorobę reumatologiczną lub nietypowym obrazem klinicznym dla IPF (np. kobieta w wieku <50 lat).
U pacjentów, u których nie stwierdza się pewnego obrazu UIP w badaniach radiologicznych lub histopatologicznych, IPF może rozpoznać zespół wielodyscyplinarny na podstawie postępującego włóknienia płuc bez innych niż IPF potencjalnych przyczyn. W tej grupie chorych wskazane jest także regularne przeprowadzanie ponownej oceny przez zespół w celu wykrycia innej przyczyny zmian płucnych (np. rozwinięcie pełnych objawów choroby tkanki łącznej) lub potwierdzenia IPF (np. uzyskanie wyniku biopsji albo ewolucja obrazu HRCT do pewnego UIP) 3 .
Problem nowotworzenia w IPF
Jednym z powodów złego rokowania w IPF oprócz postępującego charakteru choroby jest jej związek z rakiem płuca. Istnieje wiele czynników łączących te dwa procesy chorobowe, np. wywiad palenia papierosów oraz wspólne mutacje genetyczne. Mimo dużej częstości występowania raka płuca w populacji z IPF kwestia wytycznych badań przesiewowych w kierunku nowotworu podczas obserwacji pacjenta z rozpoznanym IPF nie została jak dotąd jednoznacznie rozstrzygnięta. Jest to o tyle istotne, że pewne sposoby postępowania w populacji ogólnej, np. otwarta biopsja płuca, mogą u pacjentów z IPF prowadzić do wspomnianego już nagłego zaostrzenia i w konsekwencji zgonu. Amerykańskie wytyczne National Comprehensive Cancer Network (NCCN) jako jedyne odnoszą się do kwestii IPF jako dodatkowego czynnika rozwoju raka płuca. Pacjenci, którzy przekroczyli 50 r.ż. oraz są obciążeni co najmniej 20 paczkolatami, winni być przez 3 lata rokrocznie poddawani badaniu niskodawkowej TK klatki piersiowej 5 . W przypadku populacji polskiej mamy dostępne zalecenia dla populacji ogólnej w dokumencie „Wytyczne i zalecenia zespołu ekspertów dotyczące wykrywania wczesnego raka płuca w Polsce” 6 .
Terapia
Problem terapii IPF został poruszony we wspomnianych wytycznych z 2011 r., ale w przeciwieństwie do rekomendacji dotyczących diagnostyki zaktualizowano je już w 2015 r. 7
Do czasu wprowadzenia terapii przeciwzwłóknieniowej stosowane były liczne sposoby farmakoterapii oparte na teoretycznych przesłankach dotyczących etiologii IPF. Analiza wyników wielu badań klinicznych pozwoliła sformułować rekomendacje przeciwko stosowaniu pewnych postaci farmakoterapii w IPF. I tak w wytycznych z 2015 r. rekomenduje się niestosowanie 7 :
- leczenia przeciwkrzepliwego (warfaryna) w przypadku braku innych niż IPF wskazań do jej podawania
- imatynibu
- wybiórczych antagonistów receptora endoteliny
- podwójnych antagonistów receptora endoteliny – macytentanu i bozentanu
- inhibitora fosfodiesterazy 5 – syldenafilu
- terapii skojarzonej prednizonem, azatiopryną i N-acetylocysteiną
- monoterapii N-acetylocysteiną.
Jednocześnie autorzy tych samych wytycznych warunkowo zalecają stosowanie leczenia zmniejszającego kwasowość soku żołądkowego, jak również dwóch leków przeciwzwłóknieniowych – pirfenidonu oraz nintedanibu. Pirfenidon to lek przeciwzapalny oraz przeciwzwłóknieniowy, wpływający na transformujący czynnik wzrostu β i w konsekwencji zmniejszający proliferację fibroblastów oraz produkcję kolagenu. Nintedanib jest substancją z grupy inhibitorów kinaz tyrozynowych, wpływa na działanie receptorów dla czynników wzrostu naczyniowo-śródbłonkowego, fibroblastów i czynnika płytkopochodnego.
Wszystkie trzy leki wymieniane jako warunkowo zalecane w wytycznych z 2015 r. zostały ponownie ocenione w cytowanych już rekomendacjach PTChP z 2020 r. Leczenie podwyższające pH soku żołądkowego uzyskało w tym dokumencie rekomendację negatywną. Oba leki przeciwzwłókniające uzyskały rekomendację do stosowania w IPF. Dodatkowo doprecyzowano pewne szczegóły dotyczące terapii. Nie należy stosować obu leków przeciwzwłókniających jednocześnie. Niemniej w przypadku nietolerancji lub nieakceptowalnych działań niepożądanych sugeruje się zamianę jednego leku na drugi. W Polsce pirfenidon i nintedanib są refundowane w ramach programu lekowego. Jego kryteria włączenia ograniczają grupę pacjentów, u których można stosować tę terapię. Pacjenci z ciężkimi zaburzeniami czynnościowymi płuc oraz ci, u których choroba postępuje w trakcie leczenia, powinni być wyłączani z terapii w programie. W opozycji do tych zasad stoją wytyczne PTChP sugerujące rozpoczynanie leczenia niezależnie od obecności ciężkich zaburzeń czynnościowych lub występowania jakichkolwiek zaburzeń w tym zakresie. Jednocześnie autorzy tego dokumentu nie uważają progresji choroby za czynnik warunkujący zakończenie terapii 1 .
Postępowanie niefarmakologiczne
W postępowaniu niefarmakologicznym w IPF rekomenduje się stosowanie rehabilitacji oddechowej. Powinna ona obejmować ćwiczenia fizyczne, edukację dotyczącą natury i leczenia choroby, postępowanie dietetyczne oraz wsparcie psychologiczne i socjalne. Oprócz zinstytucjonalizowanej formy rehabilitacji sugeruje się jej kontynuację w warunkach domowych.
Z uwagi na postępujący charakter choroby w jej późniejszym okresie rozwija się hipoksemiczna niewydolność oddychania. Mimo braku dowodów z randomizowanych badań klinicznych rekomenduje się, by w takich przypadkach u pacjentów z IPF stosować przewlekłą tlenoterapię domową, podobnie jak u chorych z przewlekłą obturacyjną chorobą płuc (POChP). Istnieją jednak dowody na krótszy czas przeżycia chorych z IPF w porównaniu z pacjentami z POChP. Dodatkowo w przypadku ostrej niewydolności oddychania u pacjentów z IPF nie należy stosować inwazyjnej wentylacji z uwagi na brak wpływu na czas przeżycia i rokowanie odległe. Wyjątkiem jest wentylacja inwazyjna stosowana w charakterze leczenia pomostowego w oczekiwaniu na przeszczepienie płuc. We wcześniejszym okresie choroby może dochodzić do spadków saturacji w przypadku wysiłku fizycznego. U osób, u których występuje to zjawisko, sugeruje się stosowanie tlenu podczas wysiłku 1, 7 .
W IPF, podobnie jak w innych przewlekłych chorobach układu oddechowego, rekomendowane są coroczne szczepienia przeciwko grypie oraz przeciwko Streptococcus pneumoniae 1 .
Przeszczepianie płuc
Mimo postępu zarówno w farmakoterapii, jak i w leczeniu niefarmakologicznym nie dysponujemy terapią, która mogłaby w pełni zahamować patologiczny proces prowadzący do inwalidztwa oddechowego i zgonu u pacjentów z IPF. Dlatego każdy pacjent po uzyskaniu rozpoznania, u którego nie występują przeciwwskazania, powinien być kierowany do ośrodka referencyjnego zajmującego się przeszczepianiem płuc 1, 7 . Niemniej 5 lat po przeszczepieniu płuca przeżywa około 50% pacjentów 8 .
Modyfikacja postępowania w dobie COVID-19
Opisywane wyżej postępowanie nie bierze pod uwagę zmian związanych z sytuacją pandemiczną wywołaną COVID-19. Różne międzynarodowe gremia zaproponowały zmiany w zakresie rozpoznawania i leczenia chorób śródmiąższowych w dobie pandemii. W kwestii diagnostyki proponuje się odroczenie lub rezygnację z biopsji chirurgicznej oraz wykonywania BAL z uwagi na największe ryzyko przeniesienia zakażenia SARS-CoV-2. Wszystkie możliwe konsultacje z pacjentem, jak również w ramach zespołu wielodyscyplinarnego powinny się odbywać z wykorzystaniem metod porozumiewania na odległość. Chorzy z rozpoznanym IPF leczeni antyfibrotycznie powinni kontynuować terapię. Leki antyfibrotyczne nie mają działania immunosupresyjnego, nie zwiększają ryzyka zakażenia SARS-CoV-2, ale redukują ryzyko nagłych zaostrzeń. Jeżeli w związku z pandemią konieczne jest przerwanie leczenia (chory nieprzytomny, z niewydolnością wątroby itp.), można to zrobić bezpiecznie na okres maksymalnie 4-8 tygodni. Ponowne rozpoczynanie terapii powinno odbywać się pod kontrolą ośrodka referencyjnego 9, 10 .
Podsumowanie
Idiopatyczne włóknienie płuc to najczęściej diagnozowane idiopatyczne śródmiąższowe zapalenie płuc. Złotym standardem rozpoznawania jest HRCT uzupełniana o dokładny wywiad i badania krwi ukierunkowane na wykrycie potencjalnych przyczyn wtórnego uszkodzenia układu oddechowego. W przypadku niejednoznacznego obrazu tomograficznego w diagnostyce można wykorzystać różne formy biopsji płuca i BAL. Ostateczne rozpoznanie powinno być ustalone w ramach zespołu wielodyscyplinarnego. Leczenie IPF obejmuje stosowanie terapii przeciwzwłókniającej, rehabilitację oddechową i terapię powikłań, w tym tlenoterapię domową w przewlekłej niewydolności oddechowej. Zawsze należy rozważyć skierowanie pacjenta do ośrodka transplantacyjnego.
Abstract
The diagnosis and management of idiopathic pulmonary fibrosis
Idiopathic pulmonary fibrosis (IPF) is one of the most often diagnosed interstitial lung diseases. IPF tends to affect people aged over 60 years with a history of smoking. The progressive nature of IPF leads to diminished quality of life and shorter life expectancy. Advancements in the diagnostic criteria and a greater access to high-resolution computed tomography allow an earlier and more confident diagnosis. In addition, antifibrotic treatment holds the promise of increasing the duration and quality of life. This paper discusses the present diagnostic criteria and possible treatment options.
- 1. Piotrowski WW, Bestry I, Białas AJ i wsp. Wytyczne Polskiego Towarzystwa Chorób Płuc dotyczące diagnostyki i leczenia idiopatycznego włóknienia płuc. Pneumonologia Polska 2020;1:9-67
- 2. Raghu G, Remy-Jardin M, Myers JL, et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med 2018;198:e44-e68. doi: 10.1164/rccm.201807-1255ST
- 3. Lynch DA, Sverzellati N, Travis WD, et al. Diagnostic Criteria for Idiopathic Pulmonary Fibrosis: A Fleischner Society White Paper. The Lancet Respiratory Medicine 2018;6:138-53. doi: 10.1016/S2213-2600(17)30433-2
- 4. Raghu G, Collard HR, Egan JJ, et al. An Official ATS/ERS/JRS/ALAT Statement: Idiopathic Pulmonary Fibrosis: Evidence-Based Guidelines for Diagnosis and Management. Am J Respir Crit Care Med 2011;183:788-824. doi: 10.1164/rccm.2009-040GL
- 5. Wood DE, Kazerooni EA, Baum SL, et al. Lung Cancer Screening, Version 3.2018, NCCN Clinical Practice Guidelines in Oncology. Journal of the National Comprehensive Cancer Network 2018;16:412-41. doi: 10.6004/jnccn.2018.0020
- 6. Rzyman W, Didkowska J, Dziedzic R, et al. Wytyczne i zalecenia zespołu ekspertów dotyczące wykrywania wczesnego raka płuca w Polsce. Advances in Respiratory Medicine 2018;86:50-72
- 7. Raghu G, Rochwerg B, Zhang Y, et al. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline: Treatment of Idiopathic Pulmonary Fibrosis. An Update of the 2011 Clinical Practice Guideline. Am J Respir Crit Care Med 2015;192:e3-e19. doi: 10.1164/rccm.201506-1063ST
- 8. Laporta Hernandez R, Aguilar Perez M, Lázaro Carrasco MT, et al. Lung Transplantation in Idiopathic Pulmonary Fibrosis. Med Sci (Basel) 2018;6. doi: 10.3390/medsci6030068
- 9. Wong AW, Fidler L, Marcoux V, et al. Practical Considerations for the Diagnosis and Treatment of Fibrotic Interstitial Lung Disease During the Coronavirus Disease 2019 Pandemic. Chest 2020;158:1069-78. doi: 10.1016/j.chest.2020.04.019
- 10. Spencer L. Advice for Managing Interstitial Lung Disease Patients during COVID-19 Pandemic. https://www.brit-thoracic.org.uk/media/455101/bts-management-advice-for-ild-patients-v10-23-march-2020.pdf
Następny artykuł:
Idiopatyczne włóknienie płuc – postępowanie diagnostyczno-terapeutyczne


 Dodaj do ulubionych
Dodaj do ulubionych
