


Co znajdziesz w artykule?
- Epidemiologia nowotworów ginekologicznych
- Podział nowotworów kobiecego układu płciowego pod względem histologicznym
- Molekularne mechanizmy warunkujące powstanie nowotworów złośliwych żeńskich narządów płciowych
Spis treści
Obecnie na całym świecie obserwuje się systematyczny wzrost liczby zachorowań na nowotwory złośliwe. W 2020 roku odnotowano 19,3 mln nowych przypadków, z których około 10 mln zakończyło się zgonem 1 . Należy oczekiwać, że wydłużający się czas życia człowieka oraz rozwój nauki będą prowadziły do zwiększonej zapadalności na nowotwory złośliwe, także ginekologiczne. W 2020 roku łącznie blisko 1,4 mln kobiet na świecie chorowało na nowotwory żeńskiego narządu płciowego, a niemal 675 000 zmarło
z ich powodu 1 . Przedstawione dane epidemiologiczne świadczą o tym, że nowotwory ginekologiczne stanowią wciąż poważny problem zdrowia publicznego. Do najczęściej rozpoznawanych należą rak piersi oraz rak szyjki macicy, których występowanie jest silnie związane z uwarunkowaniami socjoekonomicznymi, ze stylem życia oraz zmianami demograficznymi 2 .
W Polsce z kolei obserwuje się wzrost liczby nowych przypadków raka jajnika, raka piersi oraz raka błony śluzowej trzonu macicy (raka trzonu macicy, raka endometrium), podczas gdy zapadalność na raka szyjki macicy oraz raka pochwy się zmniejszyła. Jeżeli chodzi o raka sromu, to liczba nowych przypadków utrzymuje się na względnie stałym poziomie. Od 2010 roku zwiększa się umieralność z powodu raka piersi, podobnie jak związana z rakiem sromu oraz rakiem trzonu macicy. Z kolei umieralność na raka szyjki macicy się zmniejszyła, a w odniesieniu do raka jajnika pozostaje na stabilnym poziomie 2 .
Molekularne mechanizmy rozwoju nowotworów
Przedstawione wyżej dane statystyczne jednoznacznie wskazują na potrzebę zgłębiania znanych i poszukiwania nowych przyczyn rozwoju nowotworów ginekologicznych. Obecnie uważa się, że transformacja nowotworowa, czyli proces przemiany prawidłowej komórki w nowotworową, jest m.in. konsekwencją oddziaływania na nią czynników prowadzących do uszkodzenia jej materiału genetycznego 3 . Do powszechnie znanych mutagenów zalicza się m.in.: promieniowanie jonizujące, promieniowanie ultrafioletowe (UV), wirusy, pestycydy oraz środki bojowe. Mutacje mogą mieć charakter wrodzony, nabyty, mieszany bądź być dziełem przypadku.
W każdej komórce istnieją mechanizmy naprawy uszkodzonego materiału genetycznego. Jeżeli naprawa DNA nie jest możliwa, to następuje indukcja zaprogramowanej śmierci komórki, która zwykle odbywa się na drodze apoptozy 3 . W rozwoju nowotworów znaczenie ma także osobnicza predyspozycja genetyczna. W każdej komórce znajdują się geny odpowiedzialne za regulację jej funkcji życiowych. Zalicza się do nich protoonkogeny, które w chwili zaistnienia mutacji podlegają aktywacji do onkogenów bezpośrednio inicjujących proces transformacji nowotworowej. W przeciwieństwie do protoonkogenów geny supresorowe (antyonkogeny) działają hamująco na procesy proliferacji komórkowej oraz stabilizująco na procesy utrzymujące stabilność genetyczną komórki. Powstanie nowotworu nie jest jednak skutkiem pojedynczej mutacji, lecz jest następstwem wieloetapowego procesu zwanego kancerogenezą. Jego punktem wyjściowym jest założenie, że nowotwór jest chorobą powstającą pierwotnie na poziomie pojedynczej komórki, w której pojawiła się pierwsza mutacja. Taka komórka jest określana mianem komórki macierzystej nowotworu. W wyniku kolejnych podziałów komórkowych dochodzi do przekazywania uszkodzonego materiału genetycznego do komórek potomnych, które również nabywają kolejne, nowe mutacje. W ten sposób rozwija się klon komórek uzyskujący zdolność do nadmiernego wzrostu, proliferacji oraz unikania procesów apoptozy. Przeżycie nowotworu jest możliwe tylko w przypadku optymalnego odżywienia jego komórek. Z tego powodu w guzach obserwuje się procesy intensywnej angiogenezy prowadzące do rozwoju bogatego unaczynienia zaopatrującego je w tlen oraz substancje odżywcze. Niektóre komórki nowotworowe poprzez nabycie zdolności do migracji i przemieszczania się opuszczają miejsce pierwotnego rozwoju, a następnie drogą naczyń krwionośnych i/lub limfatycznych przedostają się do odległych miejsc organizmu, co określa się mianem przerzutowania 3 . W dziedzinie ginekologii onkologicznej molekularne podłoże chorób odgrywa istotną rolę.
Rak jajnika
Rak jajnika to jeden z najgorzej rokujących nowotworów złośliwych żeńskich narządów płciowych. W 2020 roku na świecie odnotowano 313 959 przypadków tego nowotworu, z których 207 252 zakończyły się zgonem 1 . Z kolei w Polsce w 2020 roku zarejestrowano 4669 nowych zachorowań na raka jajnika, w tym 3131 zgonów z jego powodu 4 . Szacuje się, że rak jajnika stanowi na świecie czwartą, a w Polsce trzecią przyczynę zachorowalności na nowotwory żeńskich narządów płciowych. Jest też trzecią na świecie i drugą w Polsce przyczyną umieralności z ich powodu 1, 4 .
Według danych statystycznych zaprezentowanych przez American Cancer Society 5-letnie przeżycie pacjentek z rozpoznanym inwazyjnym rakiem jajnika wynosi: 93% w przypadku choroby miejscowej, 75% w przypadku choroby obejmującej swoim zasięgiem jajnik/jajniki i regionalne węzły chłonne oraz 31% w przypadku choroby ze współistniejącymi przerzutami odległymi. Z kolei skumulowane 5-letnie przeżycie pacjentek z rakiem jajnika uwzględniające dane pochodzące z amerykańskiego rejestru Surveillance, Epidemiology and End Results (SEER) oszacowano na 49% 5 . Należy nadmienić, że statystyka w odniesieniu do raka jajnika jest zależna od typu histopatologicznego nowotworu oraz stopnia zaawansowania choroby. Dla porównania: w przypadku guzów jajnika stromalnych czy germinalnych skumulowane 5-letnie przeżycie uwzględniające SEER wynosi odpowiednio 90% i 93% 5 . W Polsce 5-letnie przeżycie chorych na raka jajnika wynosi 43,9% 6 . Efekty leczenia raka jajnika nie są satysfakcjonujące, o czym jednoznacznie świadczą przedstawione dane epidemiologiczne. Może to wynikać z kilku przyczyn, a wśród nich należy uwzględnić m.in. brak swoistej profilaktyki oraz specyfikę samego nowotworu, który przez dłuższy czas rozwija się bezobjawowo lub skąpoobjawowo. Bardzo często prowadzi to do znacznego rozwoju choroby, w związku z czym aż 75% przypadków raka jajnika jest rozpoznawanych w III lub IV stopniu zaawansowania klinicznego. W celu poprawy wyników leczenia pacjentek z rakiem jajnika w dalszym ciągu poszukuje się przyczyn molekularnego rozwoju tego nowotworu, które mogą stać się punktem docelowym nowoczesnych terapii. W piśmiennictwie dostępnych jest wiele różnych podziałów raka jajnika. Pod względem histologicznym wyróżnia się nowotwory pochodzenia nabłonkowego oraz nienabłonkowego. W tabeli 1 zaprezentowano histologiczny podział raka jajnika.
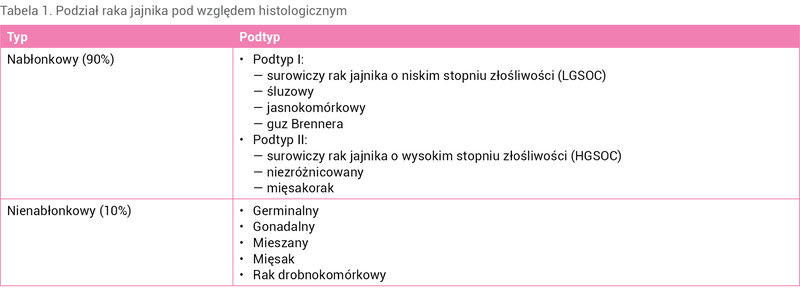
Tabela 1. Podział raka jajnika pod względem histologicznym
Najczęściej występującym typem raka jajnika jest nabłonkowy (90% przypadków), który dzieli się na podtyp I oraz podtyp II. Przedstawiony podział jest tłumaczony m.in. obserwacją, w której nowotwory podtypu I rozwijają się zwykle na podłożu endometriozy lub guzów granicznych, klinicznie charakteryzują się mniejszą złośliwością oraz są diagnozowane na wcześniejszych etapach rozwoju, podczas gdy nowotwory podtypu II rozwijają się przeważnie z komórek jajowodu, a klinicznie odznaczają się większą złośliwością i niekorzystnym rokowaniem 7, 8 . W niniejszym artykule skupiono się na raku jajnika pochodzenia nabłonkowego.
Istnieje wiele genów, których nieprawidłowości mogą skutkować rozwojem raka jajnika. W piśmiennictwie wymienia się zwłaszcza następujące: TP53, BRCA1/BRCA2, PIK3CA oraz KRAS. Częstość występowania mutacji przedstawionych genów zależy od podtypu raka. Mutacje genu TP53 dotyczą w szczególności surowiczego raka jajnika o wysokim stopniu złośliwości (HGSOC – high-grade serous ovarian carcinoma). Mutacje genów BRCA1/BRCA2 są typowe dla dziedzicznego raka jajnika oraz nawracającej postaci HGSOC. Z kolei mutacje genu PIK3CA są charakterystyczne dla raka jasnokomórkowego i endometrioidalnego. Natomiast w przypadku surowiczego raka jajnika o niskim stopniu złośliwości (LGSOC – low-grade serous ovarian carcinoma) oraz raka śluzowego często mutacje dotyczą genu KRAS 9 .
Gen TP53 koduje białko p53. Jego fizjologiczna rola sprowadza się do kontroli tzw. genów docelowych odpowiedzialnych za apoptozę, monitorowanie cyklu komórkowego oraz naprawę DNA. Białko p53 odgrywa kluczową rolę w życiu komórki, ponieważ determinuje jej przetrwanie bądź samozniszczenie. Z tego powodu stężenie białka p53 w prawidłowej komórce jest ściśle kontrolowane. Odbywa się to za pośrednictwem białka regulatorowego MDM2, które wiąże się z białkiem p53 i tworzy kompleks MDM2–p53. Następnie jest on degradowany za pośrednictwem proteasomów. Z tego powodu w zdrowych komórkach obserwuje się niewykrywalne lub niskie stężenie białka p53. W sytuacji gdy dochodzi do mutacji w genie TP53, następuje produkcja nieprawidłowego białka p53, które przestaje pełnić swoje fizjologiczne funkcje. Uszkodzone DNA – prowadzące do mutacji genu TP53 – zaburza proces łączenia się białka MDM2 z białkiem p53, co przyczynia się do zwiększenia stężenia białka p53 w komórce. Przedstawiona zależność tłumaczy także, dlaczego w komórkach nowotworowych obserwuje się zdecydowanie większe stężenia białka p53 9, 10 . Powyższy mechanizm molekularnego rozwoju raka jajnika pozwala na opracowywanie innowacyjnych metod terapii tego nowotworu. Badacze wskazują na potencjalną możliwość zablokowania nieprawidłowego białka p53 oraz indukcję apoptozy komórek nowotworowych za pośrednictwem specjalnych molekuł: PRIMA-1 lub APR-017 oraz PRIMA-1Met lub APR-246. Obecnie trwa II faza badań klinicznych nad APR-246. Środek ten został zastosowany w grupie pacjentek z nawracającym HGSOC. Na ten moment wiadomo, że lek odznacza się dobrą tolerancją, natomiast na pozostałe wyniki badania musimy jeszcze poczekać 9 . Inną cząsteczką o potencjalnym charakterze leczniczym jest RG7388 będąca inhibitorem białka regulatorowego MDM2. Zablokowanie MDM2 ma na celu uniknięcie degradacji prawidłowego białka p53, które także występuje w komórce nowotworowej. Zwiększone stężenie białka p53 sprzyja nasileniu fizjologicznych działań charakterystycznych dla prawidłowej jego formy i tym samym ogranicza progresję procesu nowotworowego 9 .
Geny BRCA1 oraz BRCA2 należą do genów supresorowych, których produkty uczestniczą w procesach naprawy DNA. Wystąpienie mutacji germinalnych omawianych genów powoduje niestabilność DNA w postaci pęknięć chromosomów, aneuploidii oraz amplifikacji centrosomów 11 . Predysponuje to do rozwoju dziedzicznego raka piersi i/lub jajnika. Wśród kobiet z rakiem jajnika 10-25% jest jednocześnie nosicielkami mutacji genów BRCA1 lub BRCA2. Prawdopodobieństwo wystąpienia raka jajnika u nosicielek mutacji w genie BRCA1 wynosi 24-54%, natomiast mutacji w genie BRCA2 – 11-27% 12 . Rozpoznanie raka jajnika BRCA-zależnego jest związane z występowaniem cech odróżniających go od sporadycznego raka jajnika, takich jak:
- średni wiek zachorowania na raka jajnika BRCA-zależnego jest mniej więcej o 5 lat niższy niż w przypadku sporadycznego raka jajnika
- wiek zachorowania na raka jajnika BRCA1-zależnego jest mniej więcej o 10 lat niższy niż w przypadku raka jajnika BRCA2-zależnego
- rak jajnika BRCA-zależny jest zwykle rozpoznawany w zaawansowanym stadium rozwoju oraz częściej odznacza się niskim stopniem zróżnicowania (G3)
- w przypadku raka jajnika BRCA1-zależnego częstość występowania nawrotów jest większa niż nawrotów raka jajnika BRCA2-zależnego 12 .
Identyfikacja pacjentek będących nosicielkami mutacji genów BRCA w odniesieniu do dziedzicznego raka jajnika ma wpływ na wdrożenie odpowiedniej profilaktyki oraz ewentualnego leczenia. U nosicielek mutacji genów BRCA można rozważyć profilaktyczną adneksektomię. Możliwe jest także zastosowanie doustnej antykoncepcji hormonalnej, choć wpływ takiego postępowania na ryzyko rozwoju raka piersi nie jest jeszcze do końca poznany 12 . W przypadku leczenia dziedzicznego raka jajnika należy zwrócić uwagę, że w porównaniu ze sporadycznym nowotworem odznacza się on lepszą odpowiedzią na chemioterapię opartą na związkach platyny oraz jest bardziej wrażliwy na terapię inhibitorami polimerazy poli-ADP-rybozy (PARP – poly-ADP-ribose polymerase) 12 . To tłumaczy, dlaczego średni czas przeżycia pacjentek z dziedzicznym rakiem jajnika jest dłuższy niż w przypadku zachorowania na sporadycznego raka jajnika 12 .
Gen PIK3CA koduje białko, które odgrywa zasadniczą rolę w regulacji szlaku PI3K/AKT/mTOR będącego złożoną ścieżką sygnalizacyjną koordynującą wiele procesów komórki, w tym: metabolizm, przeżycie oraz wzrost 13 . W przypadku raka jajnika dochodzi do zaburzenia prawidłowej funkcji tego szlaku. Zmiany liczby kopii w genach kodujących podjednostki p110α (PIK3CA) oraz p110β (PIK3CB) cząsteczki PI3K wiążą się z gorszym rokowaniem. Nadekspresja zarówno PIK3CA, jak i fosforylowanego AKT (pAKT) są związane ze zmniejszoną przeżywalnością pacjentek. Dysregulacja szlaku sygnalizacyjnego PI3K/AKT/mTOR w przebiegu raka jajnika wiąże się z uzyskiwaniem gorszych wyników leczenia 13 .
Rak błony śluzowej trzonu macicy
Rak błony śluzowej trzonu macicy to jeden z częstszych nowotworów złośliwych żeńskich narządów płciowych. W 2020 roku na świecie odnotowano 417 367 przypadków tego nowotworu i 97 370 zgonów z jego powodu 1 . Z kolei w Polsce w 2020 roku zarejestrowano 9869 nowych zachorowań na raka trzonu macicy, z których 2195 zakończyło się zgonem 4 . Szacuje się, że na świecie rak trzonu macicy stanowi trzecią przyczynę zachorowalności na nowotwory żeńskich narządów płciowych oraz czwartą przyczynę umieralności z ich powodu, a w Polsce odpowiednio: drugą i trzecią 1, 4 .
Od jakiegoś czasu liczba nowych przypadków tego nowotworu systematycznie wzrasta. Może to być związane m.in. z epidemią otyłości (liczba otyłych osób na świecie podwoiła się w ciągu niespełna 30 lat) 14 . Potwierdzeniem tego jest fakt, że 34% wszystkich przypadków raka trzonu macicy ma związek z wysokim wskaźnikiem masy ciała (BMI – body mass index) 14 . W porównaniu z kobietami z BMI <25 kg/m 2 pacjentki z BMI 30-35 kg/m 2 oraz BMI >35 kg/m 2 były narażone na odpowiednio 2,6 oraz 4,7 razy większe ryzyko zachorowania na ten nowotwór 15 . Wytłumaczeniem tego zjawiska jest m.in. znaczna zawartość estrogenów w tkance tłuszczowej, które sprzyjają rozrostom błony śluzowej macicy 16 . Poza tym tkanka tłuszczowa jest także miejscem, z którego są wydzielane adipokiny, czyli związki o charakterze prozapalnym (np. interleukina [IL] 6, leptyna), mające zdolność indukcji szlaków prowadzących do następczej kancerogenezy 16 . Ponadto otyłość może być konsekwencją nieprawidłowo zbilansowanej diety, która w dwojaki sposób wpływa na ryzyko rozwoju prezentowanego nowotworu. Dieta z dużą zawartością produktów pochodzenia zwierzęcego może zwiększać prawdopodobieństwo zachorowania na raka trzonu macicy 17 , podczas gdy dieta bogata w warzywa, owoce czy ryby może to ryzyko zmniejszać 18 . Świadczy to o udziale czynników środowiskowych w kancerogenezie raka trzonu macicy, którego rozwój jest również związany ze zmianami na poziomie DNA komórki.
W powszechnym podziale raka endometrium uwzględnia się 2 jego typy:
- typ I – endometrioidalny (EEC – endometrioid endometrial carcinoma)
- typ II – nieendometrioidalny (NEEC – non-endometrioid endometrial carcinoma).
Typ endometrioidalny jest częstszy i stanowi blisko 80% przypadków zachorowań na raka endometrium. Zwykle wykazuje niski stopień złośliwości. Dotyczy w szczególności kobiet w wieku okołomenopauzalnym ze współistniejącymi czynnikami ryzyka, takimi jak: nadciśnienie tętnicze, cukrzyca typu 2 oraz otyłość. EEC jest nowotworem estrogenozależnym, a punktem jego wyjścia są zmiany o charakterze rozrostu endometrium z atypią 19, 20 . Rak endometrium typu I na ogół występuje sporadycznie, choć w 3% przypadków jest dziedziczny i towarzyszy dziedzicznemu rakowi jelita grubego niezwiązanemu z polipowatością (HNPCC – hereditary non-polyposis colorectal cancer). W tym zespole prawdopodobieństwo pojawienia się EEC wynosi 60% i jest spowodowane mutacjami germinalnymi genów naprawy DNA (MMR – mismatch repair), takich jak: MLH1, MSH2, MSH6, PMS2 20 . Wśród innych molekularnych mechanizmów rozwoju EEC wymienia się niestabilność mikrosatelitarną (MSI – microsatellite instability) oraz mutacje w genach PTEN, KRAS, PIK3CA i CTNNB1. Wskazuje się, że MSI jest związana z hipermetylacją promotora genu MLH1 i dotyczy 75% przypadków EEC związanego z HNPCC oraz 25% przypadków EEC występującego sporadycznie. MSI poprzez zmiany w krótkich sekwencjach DNA predysponuje do zmian w różnych genach doprowadzających do rozwoju EEC 19 . U pacjentek z rakiem endometrium typu I związanym z MSI często obserwuje się współistnienie mutacji w genie PTEN. Mutacja genu PTEN dotyczy 37-61% przypadków EEC współistniejącego z MSI oraz 24-35% przypadków EEC bez MSI i jest to zarazem najczęstsza spośród mutacji dotyczących EEC 19, 20 . Podkreśla się, że w przypadku raka endometrium związanego z mutacją genu PTEN oraz z MSI zmiany nowotworowe pojawiają się już w jego zmianach prekursorowych, czyli w rozroście endometrium z atypią 20 . Często z mutacją w genie PTEN współistnieją także mutacje w genie PIK3CA, które obserwuje się u 24-39% chorych na EEC 19 . Na podstawie dostępnych danych z piśmiennictwa stwierdzono, że EEC współistniejące z mutacją w genie PIK3CA odznaczają się wysokim stopniem złośliwości oraz znaczną inwazją miometrium 20 . W rozwoju EEC nie bez znaczenia jest również mutacja w genie kodującym białko SPRY2. Fizjologicznie odpowiada ono za zmniejszenie stężenia receptora dla czynnika wzrostu fibroblastów (FGFR – fibroblast growth factor receptor). W raku endometrium typu I obserwuje się inaktywację SPRY2, co skutkuje wzrostem stężenia FGFR oraz nasilonym działaniem czynnika wzrostu fibroblastów (FGF – fibroblast growth factor) indukującego wzmożoną proliferację komórek 19 . Uważa się, że współistniejąca z EEC mutacja w genie CTNNB1 jest dobrym markerem prognostycznym, choć dane z piśmiennictwa w tym zakresie pozostają niejednoznaczne 19, 20 . Ważna jest również rola tamoksyfenu – wybiórczego modulatora receptora estrogenowego (SERM – selective estrogen receptor modulator) – stosowanego w farmakoterapii kobiet z rakiem piersi. Dostępne dane wskazują na wzrost ryzyka rozwoju raka endometrium w trakcie przyjmowania tego leku 20 . Z tego powodu pacjentki otrzymujące tamoksyfen powinny być poddawane regularnym kontrolom ginekologicznym, a w przypadku stwierdzenia przerostu endometrium w badaniu ultrasonograficznym należy rozszerzyć diagnostykę o pobranie materiału z jamy macicy do badania histopatologicznego.
Typ nieendometrioidalny jest rzadszy i stanowi około 20% przypadków zachorowań na raka endometrium. Zwykle wykazuje niski stopień zróżnicowania i agresywny przebieg. Dotyczy w szczególności kobiet w wieku pomenopauzalnym i jest diagnozowany nierzadko w zaawansowanym stadium rozwoju. NEEC jest nowotworem estrogenoniezależnym, a punktem jego wyjścia mogą być atroficzne endometrium i/lub polipy endometrialne 19, 20 . Najczęstsze mutacje występujące w NEEC to te, które dotyczą genu TP53. Stwierdza się je u 80-90% chorych na NEEC. Nadekspresja białka p53 wiąże się z niekorzystnym rokowaniem 21 . Wśród pozostałych zmian predysponujących do NEEC wymienia się utratę heterozygotyczności (LOH – loss of heterozygosity) oraz mutacje STK15, p16, kadheryny E i C-erbB2 (HER2) 20 . Wyróżnia się zwykle 2 histopatologiczne typy NEEC: raka surowiczego (USC – uterine serous carcinoma) oraz raka jasnokomórkowego (CCC – clear cell carcinoma) 20 . USC rozwija się najczęściej na podłożu śródnabłonkowego raka surowiczego (EIC – endometrial intraepithelial carcinoma), którego punktem wyjścia jest najczęściej atroficzne endometrium 20 . Współistnienie NEEC z podtypem α receptora estrogenowego oraz nadekspresją białka p53 wskazuje na zaawansowany proces kancerogenezy związany z przerzutami odległymi. Z kolei współistnienie NEEC z nadekspresją HER2 wiąże się z uzyskiwaniem przez pacjentki gorszych wyników w zakresie przeżywalności. W piśmiennictwie opisywany jest również możliwy udział mutacji genu BRCA1 w rozwoju NEEC 20 .
Przedstawiona wyżej molekularna patogeneza raka endometrium świadczy o złożoności procesu kancerogenezy w przypadku tego typu nowotworu. Poza tym część raków trzonu macicy typu II rozwija się na podłożu nowotworów typu I 20, 21 . Z powyższych względów postuluje się modyfikację klasyfikacji raka endometrium i jego podział na 4 typy:
- raka endometrium z mutacjami POLE (DNA polymerase epsilon catalytic subunit)
- raka endometrium z MSI
- raka endometrium z niskim poziomem somatycznych zmian liczby kopii DNA (somatic copy-number alterations; SCNA-low)
- raka endometrium z wysokim poziomem somatycznych zmian liczby kopii DNA (SCNA-high) 21 .
Powyższa klasyfikacja uwzględnia przede wszystkim molekularną przyczynę rozwoju raka endometrium oraz pozwala na wdrożenie optymalnego i zindywidualizowanego leczenia. Wiele mutacji jest wspólnych dla raka endometrium typu I i typu II, choć częstość ich występowania jest różna. Z tego powodu w dalszym ciągu poszukuje się możliwych patognomonicznych przyczyn rozwoju poszczególnych typów tego nowotworu.
Rak szyjki macicy
W 2020 roku na świecie odnotowano 604 127 przypadków raka szyjki macicy, z których 341 831 zakończyło się zgonem 1 . Z kolei w Polsce w 2020 roku nowotwór ten wykryto u 3862 kobiet, z których 2137 zmarło z jego powodu 4 . Szacuje się, że rak szyjki macicy stanowi na świecie drugą, a w Polsce czwartą przyczynę zarówno zachorowalności na nowotwory żeńskich narządów płciowych, jak i umieralności z ich powodu 1, 4 . Powyższe dane epidemiologiczne świadczą o tym, że mimo powszechnej profilaktyki raka szyjki macicy statystyki uwzględniające zachorowalność na niego oraz śmiertelność pacjentek z jego powodu w dalszym ciągu pozostają niesatysfakcjonujące. Należy podkreślić, że rak szyjki macicy to obecnie jedyny nowotwór żeńskich narządów płciowych, któremu można zapobiegać poprzez zastosowanie skutecznej profilaktyki opartej na:
- teście wykrywającym wirusa brodawczaka ludzkiego (HPV – human papilloma virus)
- teście połączonym (tzw. cotestingu, tj. połączeniu testu wykrywającego HPV oraz badania cytologicznego)
- badaniu cytologicznym
- szczepieniach ochronnych.
Najistotniejszą przyczyną rozwoju tego nowotworu jest przetrwała infekcja onkogennym szczepem HPV. Według danych zaprezentowanych przez World Health Organization (WHO) każdego roku diagnozuje się blisko 700 000 przypadków nowotworów spowodowanych zakażeniem HPV, z których niemal 600 000 związanych jest z rakiem szyjki macicy 22 . HPV należy do rodziny Papillomaviridae. Obecnie wyróżnia się 2 rodzaje tego wirusa w zależności od miejsca zakażenia: HPV związany z infekcją śluzówek oraz HPV związany z infekcją skóry. Zwykle wysokoonkogenne szczepy HPV (16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 66, 68) odpowiadają za zakażenia śluzówek organizmu, podczas gdy niskoonkogenne szczepy HPV (6, 11, 42, 43, 44) – za zakażenia skóry. Należy podkreślić, że 70% przypadków raka szyjki macicy oraz 50% przypadków śródnabłonkowej neoplazji dużego stopnia (HSIL – high-grade squamous intraepithelial lesion/CIN3 – cervical intraepithelial neoplasia 3) jest związanych z przetrwałym zakażeniem HPV16 lub HPV18 23 .

Tabela 2. Białka wirusa brodawczaka ludzkiego (HPV)23
Molekularne podłoże rozwoju raka szyjki macicy związanego z HPV dotyczy 8 białek tego wirusa, których ekspresja jest obligatoryjna dla właściwego jego funkcjonowania, w tym onkoproteiny E1, E2 oraz E4 są odpowiedzialne za replikację HPV, podczas gdy onkoproteiny E5, E6 oraz E7 – za kancerogenezę. W tabeli 2 przedstawiono białka HPV wraz z opisem ich znaczenia 23 .
W wyniku mikrourazów nabłonka błony śluzowej szyjki macicy HPV przedostaje się do wnętrza komórki gospodarza, w której następuje degradacja elementów wirusa. Antygeny HPV zostają wyeksponowane na powierzchni komórki za pośrednictwem głównego układu zgodności tkankowej (MHC – major histocompatibility complex) klasy I oraz II w celu aktywacji odpowiedzi immunologicznej gospodarza, w której uczestniczą m.in. limfocyty T CD8+ (limfocyty cytotoksyczne aktywowane za pośrednictwem MHC klasy I) oraz limfocyty CD4+ (limfocyty pomocnicze aktywowane za pośrednictwem MHC klasy II). Wskutek aktywacji komórek układu immunologicznego następuje inicjacja procesów zmierzających do bezpośredniej neutralizacji zakażonej komórki wraz z patogenem poprzez białka enzymatyczne bądź wzbudzenia funkcji limfocytów B, których rola jest związana z produkcją swoistych przeciwciał ukierunkowanych na neutralizację określonego antygenu. Opisany mechanizm obronny gospodarza jest skutecznie unikany przez HPV za pośrednictwem wspomnianych onkoprotein (w szczególności E6 i E7), które hamują procesy rozpoznawania antygenów wirusa przez układ immunologiczny (m.in. poprzez inhibicję MHC klasy I) oraz przyczyniają się do degradacji białka p53. Prowadzi to do przetrwałej infekcji HPV stanowiącej punkt wyjścia rozwoju śródnabłonkowej neoplazji nabłonka szyjki macicy 23 . Należy podkreślić, że ponad 90% przypadków zakażenia HPV – niezależnie od wieku pacjentki – ulega spontanicznej regresji w ciągu kolejnych 6-18 miesięcy 24 , co wskazuje na rolę układu immunologicznego w zwalczaniu wirusa. W pozostałych przypadkach rozwija się wspomniana infekcja przetrwała, która dotyczy zwłaszcza pacjentek z nieprawidłowo funkcjonującym układem immunologicznym w przebiegu zakażenia ludzkim wirusem niedoboru odporności (HIV – human immunodeficiency virus) lub po przeszczepieniu narządu 24 . W piśmiennictwie wykazano, że istnieją także predyspozycje genetyczne zwiększające ryzyko rozwoju stanów przednowotworowych oraz raka szyjki macicy. Są to m.in. polimorfizmy genów zgodności tkankowej ludzkich antygenów leukocytarnych (HLA – human leukocyte antigens). Stwierdzono, że obecność alleli HLA-DRB1*09, HLA-DRB1*15 oraz HLA-DRB1*15:02 zwiększa ryzyko rozwoju raka szyjki macicy, podczas gdy obecność alleli HLA-DRB1*13, HLA-DRB1*04:06 i HLA-DRB1*12:02 wywiera efekt ochronny 25 . Wspomniane ryzyko jest również podwyższone w przypadku polimorfizmu kodonu 72 białka p53. Jego konsekwencją jest powstanie 2 rodzajów białek p53, z których jeden rozwija się z kodonu zawierającego argininę (Arg), a drugi z kodonu zawierającego prolinę (Pro). Forma białka powstająca z kodonu z argininą jest bardziej podatna na degradację przez onkoproteinę HPV E6 i z tego powodu homozygoty Arg/Arg wiążą się ze zdecydowanie większym ryzykiem rozwoju raka szyjki macicy w porównaniu z heterozygotami Arg/Pro 26 . Ponadto mutacje w genach SHKBP1, ERBB3, CASP8 oraz TGFBR2 również predysponują do wystąpienia raka szyjki macicy 27 . Duże znaczenie w rozwoju tego nowotworu mają też nieprawidłowości dotyczące szlaków sygnałowych PI3/MAPK i/lub transformującego czynnika wzrostu typu β (TGFβ – transforming growth factor β), które stwierdzane są u 70% chorych.
Mając na względzie, że w większości przypadków rak szyjki macicy rozwija się na podłożu przetrwałej infekcji HPV, nie budzi wątpliwości konieczność jego identyfikacji u pacjentek m.in. z nieprawidłowym wynikiem badania cytologicznego. Testy wykonywane w kierunku zakażenia HPV mogą identyfikować jego DNA lub mRNA. W przypadku tych pierwszych umożliwiają one wykrywanie m.in. wysokoonkogennych szczepów HPV, takich jak HPV16 i HPV18. Wynik takiego testu pozwala na oszacowanie ryzyka rozwoju dysplazji po uzyskaniu nieprawidłowego wyniku badania cytologicznego. Test ten może być stosowany w przypadku cytologicznego rozpoznania atypowych komórek nabłonka wielowarstwowego płaskiego o nieokreślonym charakterze (ASC-US – atypical squamous cells of undetermined significance) lub śródnabłonkowej neoplazji małego stopnia (LSIL – low-grade squamous intraepithelial lesion). Istotną wartość kliniczną stanowi ujemny wynik testu, który z dużym prawdopodobieństwem wyklucza obecność HSIL/CIN3 i raka szyjki macicy, a jednocześnie oznacza, że rak nie rozwinie się w ciągu następnych 6 lat. Ujemny wynik tego badania nie wyklucza jednak zmian o charakterze HSIL/CIN2 czy LSIL/CIN1, które mogą być związane z mniej onkogennymi szczepami HPV. W grupie kobiet w wieku 25-30 lat wystarczający jest test genotypujący jedynie HPV16 i HPV18. Z kolei u pacjentek powyżej 30 roku życia zaleca się wykorzystywanie testów identyfikujących wszystkie znane onkogenne szczepy HPV 28 . Wytłumaczeniem powyższego postępowania jest obserwacja, że w grupie kobiet >30 roku życia przetrwałe zakażenie HPV utrzymuje się dłużej w porównaniu z grupą pacjentek <30 roku życia 24 .
Kolejnym badaniem opartym na molekularnym podłożu rozwoju raka szyjki macicy związanego z zakażeniem HPV jest test immunocytochemiczny CINtec PLUS, który poprzez zastosowanie odpowiednich odczynników umożliwia identyfikację ekspresji białek p16 i/lub Ki67. Białko p16 jest produkowane przez komórkę w odpowiedzi na zakażenie wysokoonkogennymi typami HPV. Natomiast białko Ki67 jest związane z nadmiernym i niekontrolowanym namnażaniem się komórek. Wykrycie p16 i/lub Ki67 wskazuje na zaburzenie cyklu komórkowego spowodowanego przetrwałą infekcją wysokoonkogennego typu HPV. Dodatni wynik testu CINtec PLUS odnotowuje się w sytuacji, gdy cytoplazma minimum jednej komórki nabłonka szyjki macicy w wyniku reakcji immunocytochemicznej barwi się na brązowy kolor (świadczy to o obecności białka p16) lub jądro komórkowe co najmniej jednej komórki nabłonka szyjki macicy w wyniku reakcji immunohistochemicznej barwi się na czerwony kolor (świadczy to o obecności białka Ki67). Jednoczesne zastosowanie odczynników wykrywających obecność białek p16/Ki67 zwiększa zarówno czułość, jak i swoistość tego testu w wykrywaniu zmian przedrakowych i raka szyjki macicy 29 .
Oznaczenie stężenia białek p16 i Ki67 może wspomóc diagnostykę nie tylko cytologiczną, lecz także histopatologiczną. W tym kontekście szczególne znaczenie przypisuje się białku p16. Jego ekspresja znacznie wzrasta w tkankach, w których proces kancerogenezy indukowany wysokoonkogennym szczepem HPV jest już zaawansowany 29 .
Znajomość molekularnych podstaw rozwoju raka szyjki macicy związanego z infekcją HPV umożliwia wdrożenie właściwej diagnostyki zakażenia, a tym samym przyczynia się do wczesnej identyfikacji wirusa i rozpoczęcia odpowiedniego leczenia. Potwierdzeniem tego są niedawno opublikowane wytyczne postępowania opartego na ryzyku w przypadku nieprawidłowych wyników testów przesiewowych w kierunku raka szyjki macicy oraz wykrycia zmian prekursorowych zaproponowane przez American Society for Colposcopy and Cervical Pathology (ASCPP). Według tych rekomendacji skrining raka szyjki macicy powinno się opierać na ocenie ryzyka jego rozwoju przy uwzględnieniu poprzednich oraz obecnych wyników testu HPV i badania cytologicznego. Istotne znaczenie w tym kontekście mają czas, jaki upłynął od ostatniego testu przesiewowego, oraz historia związana z leczeniem zmian w stopniu HSIL/CIN2+ w ciągu minionych 25 lat. Na podstawie wyżej przedstawionych czynników można określić aktualne ryzyko pojawienia się klinicznego punktu końcowego definiowanego jako HSIL/CIN3+ 30 .
Nie bez znaczenia jest rola szczepień ochronnych przeciwko HPV, które poprzez indukcję swoistej odpowiedzi immunologicznej i wytworzenie komórek pamięci zapewniają ochronę przed pierwotnym zakażeniem, redukując jednocześnie ryzyko rozwoju stanów przednowotworowych i raka szyjki macicy. Należy podkreślić, że skuteczność tych szczepionek sięga 98% (95% przedział ufności [CI – confidence interval] 86-100) w przypadku braku uprzedniej infekcji HPV oraz wynosi 44% (95% CI 26-58) u pacjentek ze stwierdzoną wcześniej infekcją HPV 24 .
Rak sromu
Rak sromu to jeden z rzadziej występujących nowotworów żeńskich narządów płciowych. W 2020 roku na świecie odnotowano 45 240 przypadków tego nowotworu, z których 17 427 zakończyło się zgonem 1 . Z kolei w Polsce w 2020 roku raka sromu rozpoznano u 761 kobiet, z których 407 zmarło z jego powodu 4 . Szacuje się, że na świecie oraz w Polsce rak sromu stanowi piątą przyczynę zarówno zachorowalności na nowotwory żeńskich narządów płciowych, jak i umieralności z ich powodu 1, 4 . W dalszym ciągu liczba dostępnych opcji terapeutycznych pozostaje niesatysfakcjonująca. Skutkiem tego jest fakt, że 5-letnie przeżycie pacjentek z zaawansowanym rakiem sromu sięga 57% w przypadku zmian zaawansowanych miejscowo oraz 17% w przypadku zmian dających przerzuty odległe 31 . W związku z tym celowe wydaje się poszukiwanie molekularnych przyczyn leżących u podłoża rozwoju raka sromu, których poznanie może stać się punktem wyjścia dla zarówno udoskonalania dostępnych metod leczenia, jak i tworzenia nowych rozwiązań terapeutycznych.
Histologicznie 80-90% wszystkich przypadków raka sromu stanowi rak płaskonabłonkowy (SCC – squamous cell carcinoma). Jego dobrze poznanymi zmianami prekursorowymi są śródnabłonkowa neoplazja sromu (VIN – vulvar intraepithelial neoplasia) typu zwykłego (VIN of usual type) oraz typu zróżnicowanego (VIN of differentiated type). Różnica między poszczególnymi typami VIN wiąże się z jednym z czynników ryzyka raka sromu, tj. zakażeniem HPV, które predysponuje do rozwoju VIN typu zwykłego, podczas gdy VIN typu zróżnicowanego nie jest związana z tą infekcją.
Szacuje się, że HPV odpowiada za 43-60% wszystkich przypadków SCC. Dla porównania infekcja HPV jest stwierdzana u blisko 98% chorych na raka szyjki macicy 31 . Istotnymi czynnikami determinującymi powyższe dane statystyczne są zarówno czułość i swoistość dostępnych testów molekularnych identyfikujących HPV w materiale biologicznym, jak i niejednorodne rozpowszechnienie tego wirusa na świecie. Szacuje się, że w populacji kobiet zamieszkujących Europę HPV odpowiada za <18% przypadków raka sromu, podczas gdy w Ameryce Północnej odsetek ten wynosi 50%, a w krajach Afryki sięga nawet 75% 32 . Dotychczasowe dane naukowe wskazują, że rozwój raka sromu związanego z HPV jest związany przede wszystkim z zakażeniem HPV16. Niemniej pozostałe wysokoonkogenne typy HPV, tj.: HPV18, HPV31, HPV33, HPV45, również odgrywają istotną rolę w rozwoju tego nowotworu. Niskoonkogenne szczepy HPV, tj. HPV6 oraz HPV11, też mogą odpowiadać za rozwój raka sromu, choć liczba danych naukowych w tym zakresie jest ograniczona 32 . HPV-zależny molekularny rozwój raka sromu jest związany z monoklonalną proliferacją komórek sromu. W przypadku zmian dysplastycznych niewielkiego stopnia wyróżnia się LSIL, której powstanie wiąże się przede wszystkim z zakażeniem HPV6 oraz HPV11. Natomiast rozwój HSIL jest związany głównie z zakażeniem HPV16. Należy podkreślić, że infekcja HPV jest stwierdzana u >80% pacjentek ze zmianami o charakterze HSIL 32 . Wirus produkuje specyficzne białka o charakterze onkogennym, tj. E6 oraz E7, których zwiększone stężenie sprzyja degradacji produktu genu TP53, czyli białka p53 stanowiącego jeden z ważniejszych czynników supresji nowotworów. Białko p53 poprzez indukcję mechanizmów naprawy uszkodzeń DNA komórki oraz jej apoptozy chroni organizm przed potencjalnym rozwojem nowotworów 32, 33 . Jego niedobór indukowany zakażeniem HPV prowadzi do zwiększonej ekspresji kolejnych wirusowych białek – p16INK4A oraz p14ARF – sprzyjających nadmiernej proliferacji zakażonych komórek. Z tego powodu u pacjentek z HPV-zależnymi nowotworami poziom białka p53 jest zwykle obniżony, podczas gdy stężenie białek p16INK4A oraz p14ARF zazwyczaj jest zwiększone 32 .
HPV-niezależny rak sromu w porównaniu z rakiem zależnym od HPV rozwija się na podłożu VIN typu zróżnicowanego, przewlekłego stanu zapalnego sromu, hiperplazji płaskonabłonkowej i/lub liszaja twardzinowego. Dotyczy przede wszystkim starszych kobiet (>50 roku życia). Molekularny mechanizm rozwoju tego nowotworu obejmuje m.in. mutacje genów TP53, PTEN oraz EGFR. Mutacja genu TP53 wiąże się ze zmniejszonym stężeniem bądź brakiem białka p53, którego niedobór – jak wspomniano wyżej – zaburza procesy naprawy uszkodzeń DNA komórki oraz jej apoptozy, stanowiąc punkt wyjścia dla kancerogenezy 33 . Z kolei mutacja genu PTEN prowadzi do utraty funkcji jego produktu, tj. białka PTEN, którego fizjologiczną funkcją w komórce jest inhibicja szlaku sygnalizacyjnego kinazy AKT prowadząca do zatrzymania podziałów komórkowych i indukcji apoptozy komórki 34 . Z kolei receptor czynnika wzrostu naskórka (EGFR – epidermal growth factor receptor) jest glikoproteiną, która odpowiada za komórkową proliferację, migrację oraz różnicowanie. Wykazano, że mutacje genu EGFR stosunkowo często dotyczą pacjentek ze stwierdzonym rakiem sromu, choć patogeneza tej zależności dotychczas nie została wyjaśniona 35 . Nierzadko w przebiegu HPV-niezależnych raków sromu obserwuje się także MSI oraz hipermetylację genu CDKN2 32 .
Molekularne mechanizmy rozwoju raka sromu mają bezpośrednie przełożenie na kliniczne aspekty profilaktyki i leczenia tego nowotworu. W prewencji pierwotnej HPV-zależnego raka sromu zastosowanie znajdują szczepionki przeciwko HPV, które zawierają wysoko oczyszczone wirusopodobne cząsteczki (VLP – viral-like particles) głównego białka L1 będącego częścią kapsydu HPV. Rolą szczepionek jest wzbudzenie odpowiedzi humoralnej organizmu prowadzącej do wytworzenia swoistych przeciwciał anty-HPV. Z kolei w przypadku raka sromu HPV-niezależnego identyfikacja mutacji genów związanych z ryzykiem rozwoju tej choroby pozwala na opracowanie nowoczesnych terapii, których celem jest m.in. pośrednia bądź bezpośrednia inaktywacja białek pośredniczących w indukcji proliferacji komórek nowotworowych. Przykładem są erlotynib oraz gefitynib – antagoniści EGFR z grupy odwracalnych inhibitorów kinazy tyrozynowej, które poprzez blokadę tego enzymu prowadzą do inaktywacji EGFR niezbędnej w proliferacji komórek. W II fazie badań klinicznych odnotowano korzyści z leczenia erlotynibem u 67% pacjentek z zaawansowanym rakiem sromu 36 . Z kolei połączenie gefitynibu z trastuzumabem zwiększało wrażliwość komórek nowotworowych na stosowaną radioterapię 37 .
Rak pochwy
Rak pochwy to najrzadziej występujący nowotwór żeńskich narządów płciowych. W 2020 roku na świecie odnotowano 17 908 przypadków tego nowotworu, z których 7995 zakończyło się zgonem 1 . Z kolei w Polsce w 2020 roku wykryto go u 108 kobiet, z których 73 zmarły 4 . Szacuje się, że na świecie, a także w Polsce rak pochwy stanowi szóstą przyczynę zarówno zachorowalności na nowotwory żeńskich narządów płciowych, jak i umieralności z ich powodu 1, 4 . Jednakże w odniesieniu do populacji polskich kobiet w ciągu ostatnich niespełna 20 lat zaobserwowano spadek zarówno liczby nowych rozpoznań (p >0,05), jak i zgonów (p <0,05) 2 . W zdecydowanej większości przypadków stwierdzenie guza pochwy jest związane z przerzutami innych nowotworów do tego narządu, takich jak: rak szyjki macicy, rak endometrium, rak jelita grubego, rak jajnika oraz rak sromu. W szczególności w pierwszej kolejności należy wykluczyć współistniejącego raka szyjki macicy oraz raka sromu 38 .
Pierwotny rak pochwy może się manifestować w postaci albo raka płaskonabłonkowego (SCC; 80% przypadków), albo gruczolakoraka (AC – adenocarcinoma; 15% przypadków). Około 5% pierwotnych guzów pochwy stanowią bardzo rzadko występujące czerniaki, chłoniaki oraz mięsaki 39 .
Podobnie jak w przypadku raka sromu nowotwory pochwy również mogą być HPV-zależne i HPV-niezależne. W przypadku raka HPV-zależnego infekcja prowadzi do zmian nabłonka pochwy określanych mianem pochwowej neoplazji śródnabłonkowej (VaIN – vaginal intraepithelial neoplasia). Szacuje się, że wirus HPV wykrywany jest u blisko 94% pacjentek z VaIN. Najczęstszym genotypem HPV odpowiedzialnym za rozwój raka pochwy jest HPV16. Z kolei w piśmiennictwie wskazuje się, że czynnikami ryzyka raka pochwy niezwiązanego z zakażeniem HPV są m.in. przewlekłe stany zapalne, stan po radioterapii oraz histerektomii 38 .
Obecnie liczba danych naukowych dotyczących molekularnego podłoża rozwoju raka pochwy jest ograniczona. Powodem tego jest najpewniej niewielkie rozpowszechnienie tego nowotworu. Niemniej w dostępnych opracowaniach na ten temat stwierdza się, że w badaniach immunocytochemicznych przeprowadzonych na fragmentach tkanek z rozpoznanym rakiem płaskonabłonkowym pochwy można zidentyfikować zwiększone stężenie EGFR oraz receptora dla czynnika wzrostu śródbłonka naczyniowego (VEGFR – vascular endothelial growth factor receptor) 38 . Przedstawione dane mogą wskazywać na kierunek przyszłych badań zmierzających do ewentualnego wyjaśnienia obserwowanych zależności.
Mięsaki trzonu macicy
Mięsaki trzonu macicy to rzadko występujące guzy. Szacuje się, że stanowią 1% wszystkich przypadków nowotworów złośliwych żeńskich narządów płciowych oraz 3-7% nowotworów złośliwych macicy 40 . Pod względem histologicznym do omawianych guzów zalicza się:
- mięsaka gładkokomórkowego (LMS – leiomyosarcoma)
- mięsaka endometrialnego wywodzącego się z podścieliska (ESS – endometrial stromal sarcoma)
- mięsaka niezróżnicowanego (UUS – undifferentiated uterine sarcoma)
- guzy mieszane:
- gruczolakomięsaka (adenosarcoma)
- mięsakoraka (carcinosarcoma).
Przedstawiona klasyfikacja obejmuje w dalszym ciągu mięsakoraki, choć aktualnie w piśmiennictwie uznaje się je raczej za odróżnicowane formy raka endometrium 41 . Carcinosarcoma występuje w 50% przypadków, LMS dotyczy 30% chorych, ESS – 15%, a UUS są stwierdzane u 5% pacjentek 41 . Mięsaki trzonu macicy mogą pojawić się w każdym wieku. Szczyt zachorowania przypada jednak na 5-6 dekadę życia 42 . Omawiane guzy mogą wykazywać dynamiczny, gwałtowny bądź powolny i wieloletni wzrost 42 . Ze względu na agresywny przebieg kliniczny oraz stosunkowo wczesny rozsiew komórek nowotworowych mięsaki trzonu macicy charakteryzują się niekorzystnym rokowaniem. Z tego powodu poszukiwanie molekularnego podłoża ich rozwoju może przyczynić się do poprawy diagnostyki i leczenia tych guzów.
Mięsakoraki składają się z dwóch komponentów złośliwych: nabłonkowego oraz mezenchymalnego. W ich przebiegu obserwuje się nadekspresję m.in.: TGFβ, białka p16, białka p53, receptora estrogenowego typu β (ERβ – estrogen receptor β) oraz VEGF 43 . Przedstawione czynniki mają zarówno pośredni, jak i bezpośredni wpływ na charakter omawianych nowotworów oraz mogą posłużyć jako wskaźniki prognostyczne. W tym aspekcie istotną rolę w patogenezie mięsakoraków odgrywa TGFβ, którego obecność indukuje proces przejścia komórek nabłonkowych w komórki mezenchymalne (EMT – epithelial to mesenchymal transition), co ma wpływ na fenotyp guza, który staje się bardziej złośliwy 43 . Z kolei nadekspresja VEGF sugeruje gorsze rokowanie, podobnie jak stwierdzenie ekspresji ERβ, której obecność wiąże się ze zwiększonym ryzykiem zgonu 43 . Obecnie trwają poszukiwania genów, których mutacje mogą predysponować do rozwoju mięsakoraków. Celem takich badań jest stworzenie immunoterapii opartej na przeciwciałach monoklonalnych, których zastosowanie umożliwiłoby inhibicję nieprawidłowych genów, co jednocześnie zmniejszałoby ryzyko sarkogenezy. Do wspomnianych genów zalicza się m.in.: EGFR, CDKN2A, MET, KRAS oraz BRAF 43 .
Mięsak gładkokomórkowy jest guzem powstającym de novo bądź rozwijającym się z uprzednio istniejącego mięśniaka macicy 43 . Utrata heterozygotyczności (LOH) na chromosomie 10 częściej dotyczy LMS niż mięśniaków macicy. Ponadto uważa się, że LOH predysponuje do sarkogenezy 43 . Dotychczas wykazano, że nadekspresja białek Ki67, p16, p53 oraz p21 wiąże się z gorszym rokowaniem w przebiegu LMS 43 . Obecnie w piśmiennictwie wskazuje się, że LMS o średnicy ≥10 cm, z indeksem mitotycznym (liczba figur mitotycznych [MF – mitotic figures] na 10 pól widzenia w dużym powiększeniu [10 HPF – 10 high-power fields]) wynoszącym ≥20 MF/10 HPF, z ekspresją Ki67 na poziomie ≥10% oraz brakiem ekspresji białka Bcl2 również sugeruje gorsze rokowanie 41 . Nadekspresja metabolizmu histydyny oraz Bcl2 wiąże się natomiast z lepszym rokowaniem 43, 44 . Znajomość molekularnych wzorców LMS pozwala zatem na prognozowanie co do przeżycia pacjentki.
W przypadku mięsaków endometrialnych wywodzących się z podścieliska istotna jest identyfikacja charakterystycznych translokacji chromosomów, które pozwalają na wyodrębnienie 2 podgrup ESS. Obecność translokacji t(7;17)(p15;q21) predysponuje do ESS niskiego stopnia (low-grade ESS), podczas gdy translokacja t(10;17)(q22;p13) jest związana z ESS wysokiego stopnia (high-grade ESS) 44 . Powyższe implikuje odmienne postępowanie terapeutyczne oraz rokowanie. Warto jednak nadmienić, że niektóre nieprawidłowości chromosomalne stwierdzane w przebiegu ESS mogą imitować LMS. Tak jest w przypadku translokacji t(X;22)(p11;p13) 44 . W celu ewentualnego różnicowania ESS i LMS można posłużyć się oznaczeniem ekspresji białka CD10. W przypadku LMS jego ekspresja wynosi jedynie około 20%, podczas gdy w przebiegu ESS sięga niemal 100% 43 .
Gruczolakomięsak jest guzem mieszanym, składającym się z komponenty nabłonkowej niezłośliwej oraz mezenchymalnej złośliwej 42 . Z tego powodu dla pacjentek z adenosarcoma charakterystyczne są guzy o mniejszym stopniu złośliwości, rzadziej naciekające miometrium, a rokowanie w ich przypadku jest korzystniejsze. Obecność adenosarcoma z sarcomatous overgrowth jest natomiast niekorzystnym czynnikiem rokowniczym 43 . W jego przebiegu immunocytochemicznie stwierdza się m.in. ekspresję białek Ki67 oraz p53. Z kolei adenosarcoma bez sarcomatous overgrowth charakteryzuje się obecnością ekspresji m.in. białka CD10 oraz ER 43 . Immunofenotyp gruczolakomięsaka pozwala na identyfikację jego podtypów, których przebieg kliniczny oraz rokowanie są odmienne.
Wnioski
Przyczynami transformacji nowotworowej żeńskich narządów płciowych mogą być zarówno narażenie na czynniki środowiskowe (np. zakażenie HPV), jak i predyspozycja genetyczna. Nierzadko obserwuje się przyczyny mieszane, gdy czynniki środowiskowe przyspieszają uwarunkowaną genetycznie predyspozycję do rozwoju nowotworu. Wiele guzów wykazuje nieprawidłowości w podobnych wewnątrzkomórkowych szlakach sygnalizacyjnych, jednak ich kliniczne przebiegi są różne. Wydaje się, że może mieć to wpływ na osiągane wyniki leczenia, które w dalszym ciągu pozostają na niesatysfakcjonującym poziomie. Z tego powodu kontynuacja badań nad molekularnym podłożem rozwoju nowotworów ginekologicznych może doprowadzić do identyfikacji nowych czynników odpowiedzialnych za ich rozwój i dzięki temu stać się jednocześnie punktem docelowym nowoczesnych terapii onkologicznych.
Abstract
Molecular basis of disease in gynaecological oncology
The incidence of cancer has been on the rise around the world. The most commonly diagnosed cancers in gynaecology include breast and cervical cancer. In Poland, there has been an increase in the incidence of ovarian cancer, breast cancer and endometrial (uterine body) cancer, while the figures for cervical and vaginal cancers have been decreasing.
Neoplastic transformation in the female reproductive organs may be due to both environmental factors (e.g., HPV infection) and a genetic predisposition. Not infrequently, the causes are mixed, i.e., environmental factors accelerate a genetic predisposition to cancer. In oncological gynaecology, the molecular basis of disease plays a significant role. Many tumours demonstrate abnormalities in similar intracellular signalling pathways, but their clinical courses differ. This appears to affect treatment outcomes. Accordingly, continued research into the molecular basis for the development of gynaecological neoplasms may lead to the identification of new factors responsible for their development that will also constitute targets for novel tumour therapies. This article discusses the molecular mechanisms underlying the development of cancers of the female reproductive system.
- 1. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2021;71(3):209-49
- 2. Piechocki M, Koziołek W, Sroka D, et al. Trends in incidence and mortality of gynecological and breast cancers in Poland (1980-2018). Clin Epidemiol 2022;14:95-114
- 3. Olszewski W. Podstawy patologii nowotworów. W: Meder J (red.). Podstawy onkologii klinicznej. Warszawa: Centrum Medyczne Kształcenia Podyplomowego, 2011:29-40
- 4. World Health Organization. Poland – global cancer observatory. https://gco.iarc.fr/today/data/factsheets/populations/616-poland-fact-sheets.pdf. Dostęp 15.07.2022
- 5. American Cancer Society. Ovarian cancer. Early detection, diagnosis and staging https://www.cancer.org/cancer/ovarian-cancer/detection-diagnosis-staging/survival-rates.html. Dostęp 15.07.2022
- 6. Religioni, U. Cancer incidence and mortality in Poland. Clin Epidemiol Glob Health 2020;8(2):329-34
- 7. Rojas V, Hirshfield KM, Ganesan S, et al. Molecular characterization of epithelial ovarian cancer: implications for diagnosis and treatment. Int J Mol Sci 2016;17(12):2113
- 8. Nowak-Markwitz E, Spaczyński M. Rak jajnika – nowe spojrzenie na pochodzenie i histogenezę. Ginekol Pol 2012;83(6):454-7
- 9. Guo T, Dong X, Xie S, et al. Cellular mechanism of gene mutations and potential therapeutic targets in ovarian cancer. Cancer Manag Res 2021;13:3081-100
- 10. Kulesza M, Dansonka-Mieszkowska A, Pieńkowska-Grela B. Napraw albo zgiń – rola białka p53 w życiu komórki. Biuletyn Polskiego Towarzystwa Onkologicznego Nowotwory 2019;4(5-6):220-31
- 11. Girolimetti G, Perrone AM, Santini D, et al. BRCA-associated ovarian cancer: from molecular genetics to risk management. Biomed Res Int 2014;2014:787143
- 12. Stawicka-Niełacna M, Markowska J. Predyspozycje genetyczne do nowotworów ginekologicznych. W: Markowska J, Mądry R (red.). Zarys ginekologii onkologicznej. Tom 1. Wyd. 3. Poznań: Termedia, 2018:63-86
- 13. Cheaib B, Auguste A, Leary A. The PI3K/Akt/mTOR pathway in ovarian cancer: therapeutic opportunities and challenges. Chin J Cancer 2015;34(1):4-16
- 14. Lortet-Tieulent J, Ferlay J, Bray F, et al. International patterns and trends in endometrial cancer incidence, 1978-2013. J Natl Cancer Inst 2018;110(4):354-61
- 15. Shaw E, Farris M, McNeil J, et al. Obesity and endometrial cancer. Recent Results Cancer Res 2016;208:107-36
- 16. McDonald ME, Bender DP. Endometrial cancer: obesity, genetics and targeted agents. Obstet Gynecol Clin North Am 2019;46(1):89-105
- 17. Shivappa N, Hébert JR, Zucchetto A, et al. Dietary inflammatory index and endometrial cancer risk in an Italian case-control study. Br J Nutr 2016;115(1):138-46
- 18. Filomeno M, Bosetti C, Bidoli E, et al. Mediterranean diet and risk of endometrial cancer: a pooled analysis of three Italian case-control studies. Br J Cancer 2015;112(11):1816-21
- 19. Matias-Guiu X, Prat J. Molecular pathology of endometrial carcinoma. Histopathology 2013;62(1):111-23
- 20. Markowska A, Pawałowska M, Korcyl M i wsp. Rak endometrium typu I i II – nowe spojrzenie na etiologię i przebieg kliniczny. Curr Gynecol Oncol 2015;13(1):5-10
- 21. Siedlecki JA, Kowalewska M. Molekularna patogeneza raka błony śluzowej trzonu macicy. W: Markowska J, Mądry R (red.). Zarys ginekologii onkologicznej. Tom 1. Wyd. 3. Poznań: Termedia, 2018:619-40
- 22. International Agency for Research on Cancer. World Health Organization. https://infogram.com/4-mar-international-hpv-awareness-day-1h1749vkv91vq6z?live. Dostęp 15.07.2022
- 23. Balasubramaniam SD, Balakrishnan V, Oon CE, et al. Key molecular events in cervical cancer development. Medicina (Kaunas) 2019;55(7):384
- 24. Crosbie EJ, Einstein MH, Franceschi S, et al. Human papillomavirus and cervical cancer. Lancet 2013;382(9895):889-99
- 25. Kamiza AB, Kamiza S, Mathew CG. HLA-DRB1 alleles and cervical cancer: a meta-analysis of 36 case-control studies. Cancer Epidemiol 2020;67:101748
- 26. Kędzia W, Rokita W, Pruski D i wsp. Epidemiologia raka szyjki macicy. W: Markowska J, Mądry R (red.). Zarys ginekologii onkologicznej. Tom 1. Wyd. 3. Poznań: Termedia, 2018:619-40
- 27. Cancer Genome Atlas Research Network. Integrated genomic and molecular characterization of cervical cancer. Nature 2017;543(16):378-84
- 28. Spaczyński M, Kotarski J, Nowak-Markwitz E i wsp. Postępowanie w przypadku nieprawidłowego wyniku przesiewowego badania cytologicznego. Rekomendacje Centralnego Ośrodka Koordynującego Populacyjny Program Profilaktyki i Wczesnego Wykrywania Raka Szyjki Macicy, Polskiego Towarzystwa Ginekologicznego, Polskiego Towarzystwa Patologów i Polskiego Towarzystwa Kolposkopii i Patofizjologii Szyjki Macicy. Ginekol Pol 2009;80:129-33
- 29. Nasierowska-Guttmejer A, Kędzia W, Wojtylak S, et al. Polish recommendations regarding diagnostics and treatment of cervical squamous intraepithelial lesions according to the CAP/ASCCP guidelines. Ginekol Pol 2016;87(9):670-6
- 30. Perkins RB, Guido RS, Castle PE i wsp. Wytyczne postępowania opartego na ryzyku w przypadku nieprawidłowych wyników testów przesiewowych w kierunku raka szyjki macicy oraz wykrycia zmian prekursorowych. Wytyczne ASCCP. Med Prakt Ginekol Położ 2020;5 [tłum. Mościcki K; konsultacja: Jach R, Trzeszcz M, Mazurec M]
- 31. Palisoul ML, Mullen MM, Feldman R, et al. Identification of molecular targets in vulvar cancers. Gynecol Oncol 2017;146(2):305-13
- 32. Rakislova N, Saco A, Sierra A, et al. Role of human papillomavirus in vulvar cancer. Adv Anat Pathol 2017;24(4):201-14
- 33. Aubrey BJ, Kelly GL, Janic A, et al. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ 2018;25(1):104-13
- 34. Nero C, Ciccarone F, Pietragalla A, et al. PTEN and gynecological cancers. Cancers (Basel) 2019;11(10):1458
- 35. Clancy AA, Spaans JN, Weberpals JI. The forgotten woman's cancer: vulvar squamous cell carcinoma (VSCC) and a targeted approach to therapy. Ann Oncol 2016;27(9):1696-705
- 36. Olawaiye AB, Cuello MA, Rogers LJ. Cancer of the vulva: 2021 update. Int J Gynaecol Obstet 2021;155(Suppl 1):7-18
- 37. Alkatout I, Schubert M, Garbrecht N, et al. Vulvar cancer: epidemiology, clinical presentation and management options. Int J Womens Health 2015;7:305-13
- 38. Skręt A, Góra T. Złośliwe guzy pochwy. W: Markowska J, Mądry R (red.). Zarys ginekologii onkologicznej. Tom 1. Wyd. 3. Poznań: Termedia, 2018:583-608
- 39. Gardner CS, Sunil J, Klopp AH, et al. Primary vaginal cancer: role of MRI in diagnosis, staging and treatment. Br J Radiol Aug;88(1052):20150033
- 40. Song Z, Wang Y, Zhang D, et al. A novel tool to predict early death in uterine sarcoma patients: a surveillance, epidemiology and end results-based study. Front Oncol 2020;10:608548
- 41. Mbatani N, Olawaiye AB, Prat J. Uterine sarcomas. Int J Gynaecol Obstet 2018;143(Suppl 2):51-8
- 42. Dańska-Bidzińska A. Mięsaki macicy. W: Markowska J, Mądry R (red.). Zarys ginekologii onkologicznej. Tom 2. Wyd. 3. Poznań: Termedia, 2018:217-27
- 43. Kobayashi H, Uekuri C, Akasaka J, et al. The biology of uterine sarcomas: a review and update. Mol Clin Oncol 2013;1(4):599-609
- 44. Bidziński M, Dańska-Bidzińska A. Biologia molekularna mięsaków macicy. W: Markowska J, Mądry R (red.). Zarys ginekologii onkologicznej. Tom 2. Wyd. 3. Poznań: Termedia, 2018:229-32
Następny artykuł:


 Dodaj do ulubionych
Dodaj do ulubionych
