


Spis treści
Powikłania neurologiczne są częste w układowych zapaleniach naczyń, toczniu rumieniowatym układowym, zespole Sjögrena oraz reumatoidalnym zapaleniu stawów. Wszystkie te choroby mają wyraźny wpływ na układ nerwowy. Problemy neurologiczne zwykle pojawiają się w przebiegu choroby układowej, ale mogą być również wczesnym objawem stanu chorobowego, który nie został jeszcze rozpoznany. Neurologiczna manifestacja może wskazywać na zaburzenia o podłożu systemowym. Kluczem do właściwego leczenia jest
ustalenie rozpoznania.
Wprowadzenie
W przebiegu chorób układowych tkanki łącznej obserwuje się różnorodne objawy neurologiczne wynikające z uszkodzenia zarówno ośrodkowego (OUN), jak i obwodowego układu nerwowego. Rzadko poprzedzają one wystąpienie pełnego zespołu objawów chorób reumatycznych (np. jak w zespole Sjögrena), jednak w znacznym stopniu wpływają na jakość życia i śmiertelność w tej grupie chorych, ponadto stanowią istotny problem kliniczny.
Do zajęcia układu nerwowego dochodzi najczęściej w takich jednostkach chorobowych, jak: różne zespoły układowych zapaleń naczyń, toczeń rumieniowaty układowy (SLE – systemic lupus erythematosus), zespół antyfosfolipidowy (APS – antiphospholipid syndrome), zespół Sjögrena, a nawet reumatoidalne zapalenie stawów (RZS). Choroby te mają podłoże autoimmunologiczne, charakteryzują się licznymi objawami klinicznymi oraz nieprawidłowościami w wynikach laboratoryjnych. Zajęcie układu nerwowego (ośrodkowego i/lub obwodowego) łączy się z reguły z cięższym przebiegiem i niekorzystnym rokowaniem. Uważa się, że przyczynami wystąpienia większości objawów neurologicznych w układowych chorobach tkanki łącznej są zamknięcie światła naczynia krwionośnego lub bezpośrednie uszkodzenie komórek nerwowych przez krążące przeciwciała.
Szybkie ustalenie rozpoznania choroby podstawowej to klucz do właściwego leczenia, a wczesne i agresywne leczenie immunosupresyjne daje gwarancję poprawy stanu neurologicznego. Zaleca się terapię skojarzoną glikokortykosteroidami oraz cyklofosfamidem lub – w lżejszych przypadkach – metotreksatem, leflunomidem lub azatiopryną.
Glikokortykosteroidy doustne stosuje się w dawce początkowej od 40 do 100 mg/24 h (1 mg/kg m.c.) w przeliczeniu na prednizon. Leczenie utrzymuje się przez 2-3 miesiące, stopniowo redukując dawkę leku do dawek podtrzymujących, aby ograniczyć działania niepożądane. W szczególnie ciężkich przypadkach, np. systemowego zapalenia naczyń, dopuszcza się podawanie dożylnych pulsów z metyloprednizolonu w dawce od 500 do 1000 mg/puls, zwykle przez 3 dni, z następczym leczeniem prednizonem w dawce 1 mg/kg m.c. Jednocześnie zaleca się cyklofosfamid we wlewach dożylnych 3 pulsy po 15 mg/kg m.c. (maks. 1,2 g) co 2 tygodnie, następnie 3-6 pulsów co 3 tygodnie lub – w tej chwili rzadko ze względu na częstsze występowanie powikłań – cyklofosfamid doustnie w dawce 100-150 mg/ 24 h. W leczeniu podtrzymującym stosuje się azatioprynę 2 mg/kg/24 h, metotreksat 20-25 mg/24 h lub leflunomid 20-30 mg/ 24 h. Leczenie należy kontynuować przez co najmniej 18 miesięcy, zwłaszcza w zapaleniach naczyń. Wczesne zaprzestanie terapii grozi nawrotem choroby. Dawki glikokortykosteroidów w terapii podtrzymującej powinny być ograniczone do maks. 10 mg/24 h lub – jeśli to możliwe – odstawione.
Pierwotne zapalenie naczyń ośrodkowego układu nerwowego
Szczególną, rzadko występującą chorobą z pogranicza neurologii i reumatologii jest pierwotne zapalenie naczyń ośrodkowego układu nerwowego (PACNS – primary angiitis of the central nervous system). Dotyczy ono średnich i drobnych naczyń mózgu, a pierwszym objawem choroby jest zwykle udar. Do innych częstych objawów zaliczają się bóle głowy, napady padaczkowe oraz encefalopatia.
Rozpoznanie PACNS można ustalić na podstawie objawów wieloogniskowego lub rozlanego uszkodzenia OUN z towarzyszącymi zmianami w płynie mózgowo-rdzeniowym oraz w badaniach obrazowych, a także w badaniu histopatologicznym mózgu lub opony miękkiej. Objawy kliniczne rozwijają się zwykle przez kilka tygodni. Nie obserwuje się wzrostu parametrów zapalenia ani objawów narządowych, natomiast u większości chorych występują nieprawidłowości w badaniu płynu mózgowo-rdzeniowego, zwykle w postaci zwiększonej liczby komórek (>5 na mm 3 ) i podwyższonego stężenia białka (>100 mg/dl) oraz często obecności prążków oligoklonalnych.
Bardzo ważną rolę w diagnostyce PACNS odgrywają badania obrazowe, takie jak tomografia komputerowa (TK) i rezonans magnetyczny (MR – magnetic resonance), które wykazują rozsiane zmiany niedokrwienne, oraz badania obrazujące ściany naczyń krwionośnych, jak: angiografia metodą rezonansu magnetycznego (angio-MR), angiografia metodą tomografii komputerowej (angio-TK) lub angiografia (arteriografia) klasyczna. W badaniach tych stwierdza się zwykle obustronne zwężenie lub zamknięcie naczyń będące wynikiem procesu zapalnego ściany naczyniowej.
W diagnostyce martwiczych zmian zapalnych naczyń w obwodowym układzie nerwowym szczególne znaczenie ma biopsja mózgu i opon mózgowo-rdzeniowych. Dotychczas nie określono jasnych zasad jej wykonania, ponadto jest ona obarczona ryzykiem uzyskania fałszywie ujemnych wyników, związanych z ogniskowością zmian i trudnościami w pobraniu materiału. Biopsję powinno się przeprowadzić po MR, uwzględniając okolice stwierdzanych zmian. Najczęściej wykonuje się ją w prawej okolicy czołowej. Badanie histopatologiczne mózgu oraz opony miękkiej wykazuje zapalenie naczyń o charakterze ziarniniakowym, martwicę włóknikową ścian naczyń oraz nacieki limfocytarne. Szczególne trudności diagnostyczne stwarzają te sytuacje kliniczne, w których obraz mózgu w MR jest prawidłowy. 1, 2, 3 Pojawienie się zmian narządowych wyklucza rozpoznanie PACNS. Tylko 14% chorych na PACNS prezentowało objawy ogólne. 4
Choroba Behçeta
Do zajęcia OUN oraz – znacznie częściej – obwodowego układu nerwowego dochodzi również w innych zespołach zapaleń naczyń. Niezwykle rzadko pierwszym symptomem choroby są objawy neurologiczne. Na ogół na początku obserwuje się objawy ogólne (gorączka, utrata wagi, osłabienie, nieprawidłowości w badaniach laboratoryjnych), a dopiero później objawy narządowe wynikające z niedokrwienia tkanek i narządów.
Rzadkim typem zapalenia naczyń różnego kalibru w naszej szerokości geograficznej jest choroba Behçeta, dotykająca głównie młodych mężczyzn. Uszkodzenie OUN występuje u 25% chorych i może wynikać po pierwsze z zapalenia tkanki mózgowej (forma miąższowa), po drugie – ze zmian w naczyniach mózgowych oraz zakrzepicy zatok żylnych (bardzo rzadko). Częściej rozwija się podostre zapalenie opon mózgowo-rdzeniowych. Objawy neurologiczne pojawiają się zwykle po 3-6 latach od wystąpienia pierwszych objawów choroby (afty i nadżerki w jamie ustnej oraz w okolicach intymnych, uveitis), znacznie częściej u chorych z ludzkim antygenem leukocytarnym (HLA – human leukocyte antigen) HLA-B51. Typowym objawem neurologicznym jest ból głowy o typie napięciowym (62%), ponadto rozdrażnienie, zmęczenie, depresja i pogorszenie funkcji poznawczych, napady drgawkowe oraz zaburzenia czucia.

Tabela 1. Kryteria rozpoznania choroby Behçeta7
Kryteria rozpoznania choroby Behçeta zebrano w tabeli 1. 7
Układowe zapalenia małych naczyń
Znacznie częściej niż w chorobie Behçeta do uszkodzenia obwodowego układu nerwowego dochodzi w przebiegu układowych zapaleń małych naczyń. Neuropatia obwodowa jest najczęstszym (i długo może być jedynym) objawem choroby. Martwicze zapalenie naczyń powoduje niedokrwienie nerwów, co prowadzi do ich uszkodzenia. Neuropatia może mieć charakter ostry lub wolno postępujący. Najczęstszą manifestacją jest wieloogniskowa mononeuropatia (ponad 60%), która dotyczy nerwu strzałkowego (89%), nerwu łydkowego (84%), nerwu piszczelowego, łokciowego oraz nerwu pośrodkowego. Druga co do częstości jest symetryczna polineuropatia obwodowa, stwierdzana u mniej niż ⅓ pacjentów. Zwykle dotyczy ona dystalnego odcinka kończyn dolnych. Rzadko występuje asymetryczna neuropatia czuciowa. Często obserwuje się też zajęcie nerwów czaszkowych, na ogół twarzowego, rzadziej III lub VI. Spośród wszystkich neuropatii obwodowych neuropatie w przebiegu chorób układowych (SVN – systemic vasculitic neuropathy) występują u 85% chorych, w tym 60% w przebiegu pierwotnych układowych zapaleń naczyń, a pozostałe 15% stanowią neuropatie idiopatyczne (NSVN – non-systemic vasculitic neuropathy). 8, 9

Tabela 3. Kryteria klasyfikacyjne* eozynofilowej ziarniniakowatości z zapaleniem naczyń11
Zwykle pierwszym objawem neuropatii jest rozlany ból (ból stanowi objaw towarzyszący u ponad 80% chorych), np. w kończynie. W dalszej kolejności następuje osłabienie uszkodzonego nerwu. Około 75% pacjentów doświadcza co najmniej jednego ostrego ataku, tylko u ok. 25% choroba ma powolny, stopniowo postępujący przebieg. Typowe jest nagłe pojawienie się niedowładów lub porażeń. Niespecyficzne objawy ogólne, takie jak: utrata wagi, ogólne osłabienie, gorączki, stwierdza się u ok. 80% chorych z neuropatią o etiologii związanej z układowym zapaleniem naczyń. Do zajęcia OUN w EGPA oraz w PAN dochodzi jedynie w ok. 5% przypadków, zwykle w trakcie trwania choroby, od 2 do 3 lat od początku objawów. Do najczęstszych manifestacji zaliczają się przemijające niedokrwienie mózgu (TIA – transient ischemic attack) i zaniewidzenie jednooczne.

Tabela 4. Kryteria klasyfikacyjne* ziarniniakowatości z zapaleniem naczyń12
Opisane wyżej dwa typy zapaleń małych naczyń krwionośnych (EGPA i GPA) zaliczane są do grupy zapaleń naczyń zależnych od przeciwciał przeciw cytoplazmie granulocytów obojętnochłonnych (neutrofilów) (ANCA – anti-neutrophil cytoplasmic antibodies). PAN rozwija się na skutek uszkodzenia naczyń przez tworzące się kompleksy immunologiczne.
Toczeń rumieniowaty układowy
SLE, RZS oraz zespół Sjögrena są trzema najczęściej występującymi chorobami reumatycznymi. U chorych nimi dotkniętych stwierdza się różne neurologiczne komplikacje, które powodują wzrost śmiertelności oraz pogarszają rokowanie, a często bywają także pierwszym objawem choroby podstawowej.
Patogeneza zmian neurologicznych w SLE pozostaje niejasna. Do najczęściej opisywanych nieprawidłowości w materiale histopatologicznym mózgu chorych na SLE należą mikroudary. Zwykle są one asymptomatyczne klinicznie, nie korelują z objawami endocarditis czy zakrzepicą. Nie znaleziono też dowodów na zapalenie naczyń mózgowych. Możliwe, że uszkodzenie systemu nerwowego wynika z bezpośredniego działania przeciwciał i cytokin zapalnych. Na przykład przeciwciała antyfosfolipidowe (APL – antiphospholipid antibodies) wpływają zarówno na waskulopatie małych naczyń, jak i zakrzepicę w dużych naczyniach. Uważa się, że przeciwciała antyneuronalne oznaczane w płynie mózgowo-rdzeniowym, szczególnie przeciwciała przeciwko podjednostce NR2 receptora N-metylo-D-asparaginianu (NMDA – N-methyl-D-aspartate), mają związek z objawami neuropsychiatrycznymi, a przeciwciała przeciwko podjednostce NR1 receptora NMDA – z zapaleniem mózgu u tych chorych. Przeciwciała antyrybosomalne początkowo były uważane za marker neuropsychiatrycznego zajęcia obwodowego układu nerwowego, lecz ich czułość wynosi 26%, a specyficzność 80%, co znacząco ogranicza ich użyteczność kliniczną. Wzrost stężenia cytokin: IL6 i interferonu łączy się ze wzrostem ryzyka psychoz i drgawek.
Postać neuropsychiatryczna tocznia występuje w 14-75% przypadków. U ok. 43% chorych objawy pojawiają się przed ustaleniem rozpoznania SLE, a u 63% – w pierwszym roku od rozpoznania. Do najczęstszych zaburzeń neuropsychiatrycznych w SLE należą: bóle głowy (28-57%), zaburzenia nastroju (21%), dysfunkcje poznawcze (20%), drgawki (10%) i choroby naczyń mózgowych (8%). Chociaż bóle głowy stanowią najczęstszy objaw neurologiczny w toczniu, nie są częstsze niż w populacji ogólnej. Podobnie depresja w toczniu występuje równie często jak w populacji ludzi zdrowych.
Psychozy są rzadsze (ok. 11%) i związane z aktywnością choroby. Zwykle obejmują omamy i myśli paranoiczne. Psychozy mogą być pierwszym objawem choroby. Znacznie częściej niż psychozy obserwuje się u tych chorych zaburzenia funkcji poznawczych. W standardowych testach neuropsychiatrycznych, które stosowano w wielu dużych badaniach klinicznych, stwierdzono je u 60-80% pacjentów. Zwykle zaburzone są umiejętności wzrokowo-przestrzenne (np. rozpoznawanie kształtów, ocena odległości) oraz pamięć wzrokowa i werbalna. Obecność przeciwciał przeciwko receptorowi NMDA i przeciwciał antykardiolipinowych (ACL – anticardiolipin antibodies) w klasie immunoglobuliny M (IgM) oraz wysokie miano przeciwciał przeciwjądrowych (ANA – antinuclear antibodies) wiążą się z większym ryzykiem występowania zaburzeń poznawczych.
Drgawki mogą pojawiać się zarówno przed rozwojem innych objawów choroby, jak i podczas jej trwania. Czynniki predysponujące do drgawek to przebyty udar niedokrwienny, obecność antykoagulantu toczniowego (LA – lupus anticoagulant), APL i przeciwciał anty-Sm. Zwykle mamy do czynienia z pojedynczymi epizodami napadów drgawkowych, ale wysoka aktywność choroby predysponuje do nawracających i przedłużających się stanów drgawkowych. W SLE częściej niż w populacji ogólnej obserwowano udary niedokrwienne. Oprócz konwencjonalnych czynników ryzyka udaru (nadciśnienie, dyslipidemia, cukrzyca) – częstych u chorych na SLE, wynikających w dużej mierze z przedłużającego się leczenia glikokortykosteroidami oraz prowadzących do przyspieszonej miażdżycy – predykatorem występowania udaru są także APL. U 65% chorych na SLE z udarem stwierdzono występowanie tych przeciwciał. 14, 15, 16
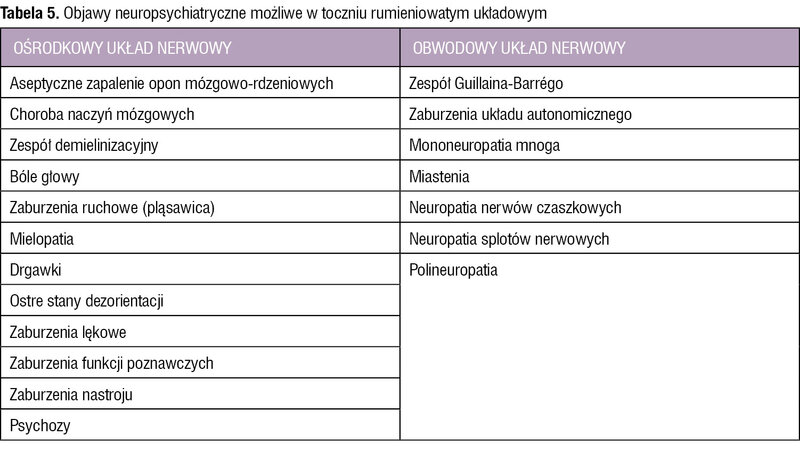
Tabela 5. Objawy neuropsychiatryczne możliwe w toczniu rumieniowatym układowym
Zespół antyfosfolipidowy
Badania wykazały, że u 50% chorych na SLE, u których stwierdzono obecność ACL, w ciągu 10 lat rozwija się wtórny APS. Wiąże się to z ryzykiem powikłań zakrzepowych i wpływa na wzrost śmiertelności w tej grupie chorych. APS może być pierwotny lub wtórny do innych chorób, w jego przebiegu występują zakrzepica w naczyniach żylnych i/lub tętniczych oraz powikłania w trakcie ciąży, którym zawsze towarzyszy obecność APL. APL stwierdzane są u ok. 1-3% dorosłej populacji bez klinicznych objawów APS, u 25% chorych z zakrzepicą żylną, u 25% młodych chorych z udarem, u 18% chorych z przedwczesną miażdżycą naczyń wieńcowych oraz u ok. 30% pacjentek z nawracającymi utratami ciąż. Udary niedokrwienne w przebiegu APS dotykają częściej młodszych pacjentów w porównaniu z tymi bez APS. Często zgłaszanym objawem są migrenowe bóle głowy, ponadto opisywano upośledzenie funkcji poznawczych (od łagodnych do ciężkich zespołów otępiennych pochodzenia naczyniowego) oraz napady drgawkowe, które ściśle wiążą się z obecnością APL. U niektórych pacjentów z APS występują objawy przypominające stwardnienie rozsiane (pseudo-SM; SM – sclerosis multiplex). U tych chorych częściej zdarzają się ogniskowe epizody niedokrwienne w mózgu: przeważnie TIA, rzadziej (zwykle u dzieci i młodych kobiet) obserwowano pląsawicę, mielopatię poprzeczną czy objawy ze strony narządu wzroku.
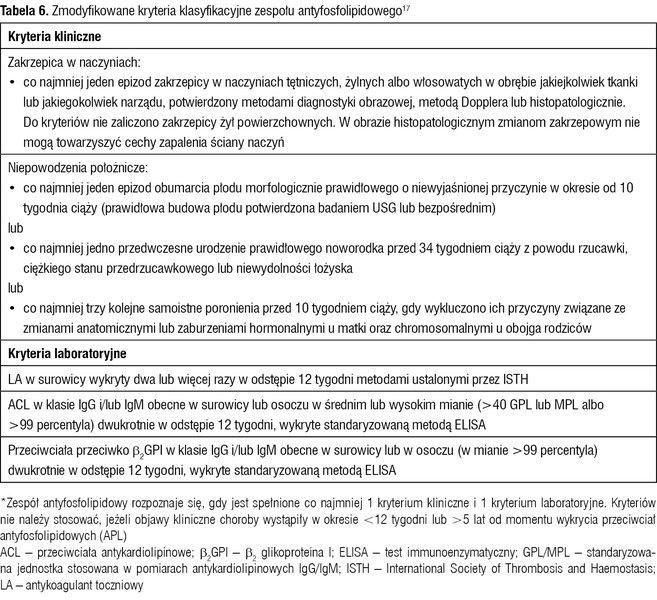
Tabela 6. Zmodyfikowane kryteria klasyfikacyjne zespołu antyfosfolipidowego17
Zespół Sjögrena
Zespół Sjögrena uważany jest za drugą co do częstości występowania po RZS chorobę autoimmunologiczną. Szacuje się, że cierpi na niego 0,5-5% populacji ogólnej, z czego ponad 90% to kobiety w wieku okołomenopauzalnym. Zespół Sjögrena może dawać wiele różnych objawów, które dzielą się na dwie duże grupy: objawy związane z zajęciem gruczołów wydzielania zewnętrznego (zespół suchości) oraz objawy związane z zajęciem innych narządów i układów.
Objawy neurologiczne, obserwowane u ok. 20% pacjentów, często są pierwszym symptomem choroby lub dominują w obrazie klinicznym. Poprzedzają wystąpienie zespołu Sjögrena u ok. 81% pacjentów czasem o 3-5 lat. Co więcej, tylko 21% chorych w momencie wystąpienia objawów neurologicznych ma typowe ANA (Ro i La), które rozwijają się w ciągu 7 następnych lat.
Zwykle u chorych obserwuje się bóle głowy (47%), zaburzenia funkcji poznawczych (44%) i zaburzenia nastroju (38%). Najczęstszym rodzajem bólu głowy jest migrena bez aury. Ponadto pacjenci często zgłaszają zaburzenia koncentracji i pamięci, w porównaniu z populacją ogólną oraz chorymi na RZS wykazują też większą skłonność do zaburzeń depresyjnych i lękowych.
Z piśmiennictwa wynika, że neuropatia w zespole Sjögrena występuje z różną częstością. W jednym badaniu, które obejmowało grupę ponad 1000 pacjentów, stwierdzono ją jedynie u 11% chorych, ale w innym badaniu 58% pacjentów bez objawów neuropatii w badaniu elektrofizjologicznym prezentowało zaburzenia typowe dla neuropatii. Neuropatia poprzedza wystąpienie zespołu Sjögrena w ok. 35% przypadków. Neuropatie mają zwykle charakter czuciowy i wynikają z uszkodzenia zwojów nerwowych korzeni czuciowych rdzenia (ganglionopatia sensoryczna) lub małych, niemielinowych włókien czuciowych (bolesna neuropatia małych włókien), ale mogą wystąpić także mononeuropatia mnoga, poliradikulopatia czy neuropatia nerwu układu autonomicznego. Ganglionopatia objawia się zwykle jako zaburzenie czucia, niestabilność chodu oraz trudności w wykonywaniu precyzyjnych ruchów. 18, 19, 20
Reumatoidalne zapalenie stawów
RZS dotyczy głównie młodych kobiet, ale istnieje także postać o późnym początku, stwierdzana u osób po 60 r.ż. Najpoważniejszym neurologicznym powikłaniem RZS jest zapalenie stawów kręgosłupa szyjnego w zakresie łącza szczytowo-potylicznego z następczą destrukcją i podwichnięciem w stawie szczytowo-obrotowym. Zmiany zapalne w odcinku C1-C2 obserwowane są u 25-80% chorych na RZS, a ich objawem jest ból promieniujący na potylicę. W razie ucisku na rdzeń kręgowy może pojawić się niedowład spastyczny kończyn górnych. Powikłanie to występuje u chorych z agresywnym przebiegiem choroby i koreluje z postępem zmian destrukcyjnych w stawach obwodowych.
Konieczne jest wykonanie rentgenogramu (RTG) w projekcji bocznej, przy głowie przygiętej do klatki piersiowej. Wówczas można uwidocznić szczelinę (3 mm) między zębem obrotnika a łukiem kręgu szczytowego.
W RZS często występuje zespół kanału nadgarstka (CTS – carpal tunnel syndrome). Tętnice odżywiające nerw są uciskane przez ścięgna pogrubione na skutek zapalenia pochewek. Częstość CTS jest wyższa u chorych z RZS niż w populacji ogólnej, szczególnie u pacjentów z dodatkowymi czynnikami ryzyka. Jest to symptom późny, pojawiający się u osób z zaawansowanym, długo trwającym procesem chorobowym. Objawia się bólem i drętwieniem palców (kciuk, wskazujący, środkowy i część serdecznego), dolegliwości wybudzają ze snu. Na początku pierwszego stadium choroby zdarza się to rzadko. Potem dolegliwości pojawiają się wielokrotnie w ciągu nocy, a ból promieniuje do przedramienia, a nawet barku. W drugim stadium drętwienie i ból występują także w dzień, zwłaszcza podczas wysiłku (np. podczas jazdy na rowerze). Towarzyszy temu pogorszenie się sprawności manualnej w przypadku czynności wymagających precyzji. W trzecim stadium CTS objawy się nasilają i pojawiają się zaniki mięśniowe. 21
Podsumowanie
Uszkodzenie układu nerwowego – zarówno mózgu i rdzenia, jak i obwodowego układu nerwowego – jest częste w chorobach układowych tkanki łącznej. Bywa, że manifestacje neurologiczne poprzedzają wystąpienie innych symptomów choroby, często są one objawem późnym lub świadczą o agresywnym przebiegu choroby. Zajęcie układu nerwowego zawsze jest objawem złym rokowniczo i wymaga intensywnego, wczesnego leczenia immunosupresyjnego.
W zapaleniach małych naczyń zwykle dochodzi do uszkodzenia obwodowego układu nerwowego. Najczęściej obserwuje się zajęcie nerwów obwodowych kończyn dolnych, znacznie rzadziej kończyn górnych. Neuropatia ma przeważnie charakter mieszany, czuciowo-ruchowy. W elektromiografii stwierdza się aksonalne uszkodzenie nerwów, często bardziej rozległe, niż sugerują to objawy kliniczne.
Do zajęcia OUN dochodzi dużo rzadziej i zwykle późno w przebiegu choroby. Objawy to: krwawienie mózgowe lub podpajęczynówkowe, udar niedokrwienny, napady drgawkowe, porażenie nerwów czaszkowych. W innych chorobach tkanki łącznej, takich jak SLE i zespół Sjögrena, zdarza się, że objawy uszkodzenia układu nerwowego poprzedzają wystąpienie pozostałych symptomów choroby. Mogą mieć one charakter pierwotny i wynikać z aktywnego procesu zapalnego, być wtórne do np. powikłań zakrzepowo-zatorowych albo stanowić skutek agresywnego leczenia. Zarówno w SLE, jak i zespole Sjögrena najczęściej stwierdza się bóle głowy, drugie w kolejności są zaburzenia funkcji poznawczych. Dla APS typowym objawem jest udar. Najgroźniejszym powikłaniem RZS pozostaje podwichnięcie w stawie szczytowo-obrotowym, które grozi wgłobieniem.
Abstract
Neurologic problems as the initial manifestation of rheumatic disease
Neurologic complications are frequent in systemic vasculitis, systemic lupus erythematosus, Sjögren syndrome and rheumatoid arthritis. Each of these systemic inflammatory diseases distinctly affects the nervous system. Neurological problems are common in the course of systemic diseases, but may also be an early symptom of a medical condition that has not yet been diagnosed. Consequently, neurologists need to be aware when a neurological presentation might indicate an underlying systemic disorder. The key to appropriate treatment is to establish the correct diagnosis.
- 1. Younger DS. Vasculitis of the nervous sytem. Curr Opin Neurol 2004;17:317-36.
- 2. Elbers J, Halliday W, Hawkins C, et al. Brain biopsy in children with primary small-vessel central nervous system vasculitis. Ann Neurol 2010;68:602-10.
- 3. McKinney JS, Cucchiara BL. Diagnosis and management of cerebral vasculitis. In: Hurst RW, Rosenwasser RH. Neurointerventional management: diagnosis and treatment. New York: Informa Healthcare, 2012.
- 4. Salvarani C, Brown RD Jr, Calamia KT, et al. Primary central nervous system vasculitis: analysis of 101 patients. Ann Neurol 2007;62:442-51.
- 5. Noel N, Bernard R, Wechsler B, et al. Long-term outcome of neuro-Behçet’s disease. Arthritis Rheumatol 2014;66:1306-14.
- 6. Lee SH, Yoon PH, Park SJ, et al. MRI findings in neuro-Behçet’s disease. Clin Radiol 2001;56:485-94.
- 7. International Study Group for Behçet’s disease. Criteria for diagnosis of Behçet’s disease. Lancet 1990;335:1078-80.
- 8. Mathew L, Talbot K, Love S, et al. Treatment of vasculitic peripheral neuropathy: a retrospective analysis of outcome. QJM 2007;100:41-51.
- 9. Blaes F. Diagnosis and therapeutic options for peripheral vasculitic neuropathy. Ther Adv Musculoskel Dis 2015;7:45-55.
- 10. Lightfoot RW Jr, Michel BA, Bloch DA, et al. The American College of Rheumatology 1990 criteria for the classification of polyarteritis nodosa. Arthritis Rheum 1990;33:1088-95.
- 11. Masi AT, Hunder GG, Lie JT, et al. The American College of Rheumatology 1990 criteria for the classification of Churg-Strauss syndrome (allergic granulomatosis and angiitis). Arthritis Rheum 1990;33:1094-100.
- 12. Leavitt RY, Fauci AS, Bloch DA, et al. The American College of Rheumatology 1990 criteria for the classification of Wegener’s granulomatosis. Arthritis Rheum 1990;33:1101-7.
- 13. Nadeau SE. Neurologic manifestations of systemic vasculitis. Neurol Clin 2002;20:123-50.
- 14. Voss E, Stangel M. Nervous system involement in connective tissue disease: mechanism and diagnostic approach. Curr Opin Neurol 2012;25:306-15.
- 15. Bhattacharyya S, Helfgott SM. Neurologic complications of systemic lupus erythematosus, Sjögren syndrome, and rheumatoid arthritis. Semin Neurol 2014;34:425-36.
- 16. Lefèvre G, Zéphir H, Warembourg F, et al. Neuropsychiatric systemic lupus erythematosus (1st part). Cases definitions and diagnosis and treatment of central nervous system and psychiatric manifestations of systemic lupus erythematosus. Rev Med Interne 2012;33:491-502.
- 17. Miyakis SI, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006;4:295-306.
- 18. Ialande S, De Seze J, Fauchais AL, et al. Neurologic manifestations in primary Sjögren syndrome: a study of 82 patients. Medicine (Baltimore) 2004;83:280-91.
- 19. Ramos-Casals M, Solans R, Rosas J, et al. Primary Sjögren syndrome in Spain: clinical and immunologic expression in 1010 patients. Medicine (Baltimore) 2009;87:210-9.
- 20. Gøransson LG, Herigstad A, Tjensvoll AB, et al. Peripheral neuropathy in primary Sjögren syndrome: a population-based study. Arch Neurol 2006;63: 1612-5.
- 21. Karadag O, Kalyoncu U, Akdogan A, et al. Ultrasonograficzna ocena zespołu cieśni nadgarstka u chorych z RZS: częstość występowania i korelacja ze stopniem ciężkości choroby. Serwis Reumatologia, 2 listopada 2012.
Następny artykuł:
Objawy neurologiczne jako pierwsza manifestacja choroby reumatycznej


 Dodaj do ulubionych
Dodaj do ulubionych
