


Co znajdziesz w artykule?
NNH (PNH – paroxysmal nocturnal hemoglobinuria) to rodzaj rzadkiej niedokrwistości hemolitycznej, która klinicznie manifestuje się hemolizą wewnątrznaczyniową, różnego stopnia niewydolnością szpiku oraz tendencją do żylnej i tętniczej zakrzepicy.[1] Jest wynikiem niezłośliwego klonalnego rozrostu wielopotencjalnych komórek krwiotwórczych (WKK), które nabyły defekt syntezy kotwicy glikozylofosfatydyloinozytolowej (GPI) we własnej błonie komórkowej. Defekt ten prowadzi do braku wielu białek powierzchniowych na WKK, m.in. odpowiedzialnych za ochronę własnych komórek organizmu przed układem dopełniacza. Choć defekt dotyczy wszystkich komórek krwi, największe znaczenie kliniczne ma hemoliza erytrocytów spowodowana niekontrolowaną aktywnością dopełniacza. NNH często występuje z niewydolnością szpiku, głównie z anemią aplastyczną. Choroba najczęściej dotyka młodych dorosłych – mediana zachorowania w populacji dorosłych wynosi 30 lat. NNH może mieć ciężki przebieg kliniczny, a 5-letnie przeżycie wynosi 35 proc. Zakrzepica jest główną przyczyną zgonów w przebiegu NNH.[1] W Polsce co roku diagnozuje się ok. 20 nowych przypadków NNH. Szacowana częstość występowania NNH na świecie to 1,3/milion/rok, co wskazuje na znaczne niedodiagnozowanie NNH w naszym kraju.[1] Dlatego ważne jest zwiększanie świadomości lekarzy na temat tego schorzenia oraz częstsze kierowanie chorych na badania.
Spis treści
PATOGENEZA
Układ dopełniacza (UD)
Układ dopełniacza jest bardzo ważnym elementem obrony przeciwzakaźnej i razem z komórkami żernymi należy do najstarszych ewolucyjnie mechanizmów odporności organizmu. 1 Obejmuje ponad 60 białek błonowych i rozpuszczalnych, które są aktywowane w określonej kolejności poprzez reakcje łańcuchowe proteaz.
Istnieją trzy drogi aktywacji dopełniacza (ryc. 1):
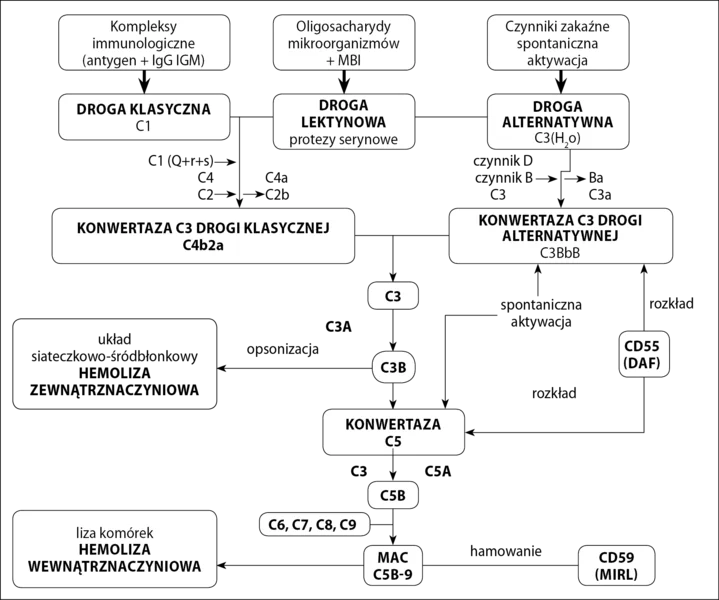
Ryc. 1. Drogi aktywacji i mechanizm działania układu dopełniacza
- klasyczna,
- lektynowa,
- alternatywna.
Wszystkie
trzy prowadzą do produkcji konwertazy C3. Konwertaza C3 jest odpowiedzialna za rozpad C3 na składową C3a i C3b. C3a pełni funkcję anafilotoksyny, a C3b łączy się z błoną komórki docelowej, tworząc konwertazę C5, która rozkłada C5 na składowe C5a i C5b. C5b przyłącza kolejno składniki C6, C7, C8. W wyniku przyłączenia cząsteczek C9 do kompleksu C5b678 powstaje kompleks atakujący błonę (MAC – membrane attacking complex), prowadzący do lizy komórki. Inicjacja drogi klasycznej i lektynowej wymaga odpowiednio połączonych z antygenem przeciwciał klasy IgG lub IgM, lub białka wiążącego mannozę związanego z oligosacharydami na powierzchni mikroorganizmów. Na drodze alternatywnej zarówno składowa C3, jak i konwertaza C3 drogi alternatywnej mają zdolność do samoistnej aktywacji.
Rola UD w patogenezie NNH została potwierdzona w 1954 roku przy okazji odkrycia alternatywnej drogi aktywacji dopełniacza. Odpowiada ona za 80-90 proc. całej aktywności dopełniacza, 1 dlatego niezbędne są mechanizmy regulujące jego aktywność, które zabezpieczają komórki własne organizmu przed jego spontanicznym działaniem. Z punktu widzenia patogenezy NNH najważniejsze błonowe czynniki regulujące UD to: CR1 (kofaktor dla czynnika I rozkładającego C4b i C3b), CD55 (DAF – decay accelerating factor – przyspiesza rozkład konwertazy C3 i C5) i CD59 (MIRL – membrane inhibitor of reactive lysis – wiąże C8 i C9 i hamuje formowanie MAC).
CD55 i CD59 są umocowane w błonie przez kotwicę z GPI (ryc. 2), co umożliwia im większą ruchomość w błonie komórkowej i efektywniejsze działanie. Zaburzenia w syntezie kotwicy GPI powodują brak/zmniejszenie ekspresji CD55 i CD59 na powierzchni komórek potomnych WKK z defektem syntezy kotwicy (erytrocyty, trombocyty, monocyty i granulocyty) oraz spontaniczną niekontrolowaną aktywację układu dopełniacza. Jedynym błonowym regulatorem dopełniacza w komórkach NNH pozostaje CR1. Brak CD59 skutkuje niekontrolowanym powstawaniem MAC i w konsekwencji hemolizą wewnątrznaczyniową. Brak CD55 prowadzi do zwiększonej opsonizacji erytrocytów przez fragmenty dopełniacza, głównie C3b. Opłaszczone erytrocyty są wychwytywane przez układ siateczkowo-śródbłonkowy i niszczone w mechanizmie hemolizy zewnątrznaczyniowej. U nieleczonych pacjentów w obrazie klinicznym dominują jednak objawy hemolizy wewnątrznaczyniowej, które maskują toczący się równolegle proces hemolizy zewnątrznaczyniowej. 1
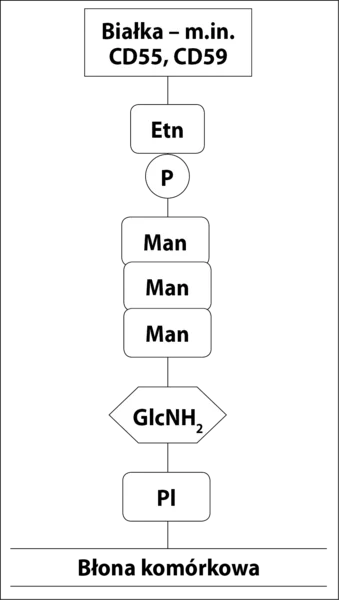
Ryc. 2. Budowa kotwicy GPI. Kotwica jest zbudowana z połączonego z błoną komórkową fosfatydyloinozytolu (PI), glukozaminy (GlcNH2), trzech cząsteczek mannozy (Man) i fosfoetanolaminy (PEtn).
Genetyka
W latach 90. XX wieku zidentyfikowano mutację genu PIG-A (Phosphatidylinositolglycan class A), która odpowiada za defekt syntezy kotwicy GPI. 1 NNH jest spowodowana nabytą (somatyczną) mutacją genu PIG-A, zlokalizowanym na Xp22 (ryc. 3). To jedyna niedokrwistość hemolityczna zależna od nabytej mutacji somatycznej komórek macierzystych szpiku 1 oraz pierwszy poznany przykład schorzenia nabytego, związanego z chromosomem X. 1 Produkt genu PIG-A odpowiada za wczesne etapy syntezy kotwicy GPI, która zachodzi posttranslacyjnie w siateczce endoplazmatycznej. Mutacja PIG-A skutkuje utratą funkcji genu, a w konsekwencji brakiem kotwicy GPI w błonie komórkowej i brakiem wielu białek powierzchniowych kotwiczonych za jej pomocą. PIG-A to jedyny zidentyfikowany dotychczas gen odpowiedzialny za fenotyp NNH (defekt GPI).

Ryc. 3 Patogeneza NNH.
Z nabywania mutacji przez WKK oraz lokalizacji genu PIG-A na chromosomie X wynika 9 :
1. pojedyncza mutacja wystarczy, aby spowodować fenotyp NNH, ponieważ komórki somatyczne są haploidalne względem genów zlokalizowanych na chromosomie X (wynika to z inaktywacji jednego chromosomu X u kobiet);
2. mozaikowatość WKK, czyli obecność różnych populacji w odniesieniu do obecności kotwicy GPI na ich powierzchni;
3. równa częstość występowania NNH u kobiet i mężczyzn; mutacja PIG-A zachodzi po inaktywacji chromosomu X, a mutacje w inaktywowanym chromosomie pozostają klinicznie nieme;
4. ekspansja komórek z defektem w syntezie kotwicy GPI.
Komórki z mutacją w genie PIG-A można znaleźć u nieznacznej liczby niechorujących na NNH 1 , czyli osób zdrowych. Mutacja PIG-A jest warunkiem koniecznym, jednak niewystarczającym do wywołania NNH. Choroba ujawnia się klinicznie wówczas, gdy komórki o fenotypie NNH zdobywają zdolność do rozrostu. Mechanizm, w jakim komórki z defektem kotwicy GPI zyskują zwiększoną przeżywalność w stosunku do innych komórek szpiku, jest przedmiotem badań. Jedna hipoteza zakłada związek ujawnienia NNH z procesem autoimmunizacyjnym, w którym komórki NNH są mniej podatne na atak komórek NK oraz mniej wrażliwe na INF-γ i TNF-α. Druga hipoteza wyjaśnia przewagę klonu z defektem GPI jako wynik drugiej mutacji (zachodzącej po mutacji PIG-A), związanej ze złośliwymi rozrostami WWK, która umożliwia im klonalny rozrost. 1
KLASYFIKACJA I DIAGNOSTYKA
The International PNH Interest Group (PNH IG) dzieli NNH na trzy kategorie:
1. Klasyczna NNH
W obrazie klinicznym występują objawy związane z hemolizą wewnątrznaczyniową. Nie występują inne nieprawidłowości w szpiku kostnym.
2. NNH w przebiegu innej wady szpiku
Oprócz hemolizy wewnątrznaczyniowej w szpiku wykrywa się anemię aplastyczną, zespół niedysplastyczny, inne mielopatie (np. mielofibrozę).
3. Subkliniczna NNH
Brak klinicznych i laboratoryjnych objawów hemolizy. Wykrywa się niewielkie populacje komórek o fenotypie NNH. Często występuje w związku z niewydolnością szpiku, w szczególności z anemią aplastyczną i zespołem mielodysplastycznym z dysplazją w układzie erytrocytów dawniej zwanym niedokrwistością oporną na leczenie (ang. refractory anemia myelodysplastic syndrome – MDS-RA)
KRYTERIA DIAGNOSTYCZNE
Aby rozpoznać oraz skategoryzować NNH, należy wykonać analizę markerów hemolizy, rozmaz krwi obwodowej, cytometrię przepływową (w celu identyfikacji komórek z defektem kotwicy GPI) oraz analizę szpiku.
Markery hemolizy
Hemoliza oznacza rozpad erytrocytów, co przejawia się ich skróconym czasem przeżycia. W NNH jest ona spowodowana niekontrolowaną aktywacją układu dopełniacza. W organizmie chorych zachodzą zarówno hemoliza wewnątrznaczyniowa (formowanie MAC w błonie erytrocytów), jak i zewnątrznaczyniowa (opsonizacja przez C3b i fagocytoza w układzie siateczkowo-śródbłonkowym), jednak u nieleczonych pacjentów dominuje obraz hemolizy wewnątrznaczyniowej. Objawy hemolizy zewnątrznaczyniowej mogą się ujawniać u pacjentów leczonych ekulizumabem. 1 Na toczący się proces hemolizy mogą wskazywać następujące markery:
1. Hemoglobina (Hb) – stężenie wolnej Hb w surowicy to najbardziej bezpośredni wskaźnik nasilenia hemolizy. 1 Nagłe zmniejszenie stężenia Hb jest charakterystyczne dla hemolizy wewnątrznaczyniowej, w której szybkość niszczenia erytrocytów jest szybsza niż w hemolizie zewnątrznaczyniowej.
2. Retikulocyty – bezpośrednie prekursory erytrocytów. Zwiększenie ich liczby odpowiada wyrównawczej odnowie linii erytroidalnej. Są wskaźnikiem aktywności szpiku. Wyrównawcza retikulocytoza może być nieobecna w przypadku współistniejącej niewydolności szpiku. Na dysfunkcje szpiku wskazują również towarzyszące neutropenia i trombocytopenia.
3. Dehydrogenaza mleczanowa (LDH, lactate dehydrogenase) – enzym uwalniany ze zhemolizowanych erytrocytów. Hemolizę wewnątrznaczyniową cechuje większy wzrost stężenia LDH niż zewnątrznaczyniową. Stężenie koreluje dodatnio z nasileniem hemolizy.
4. Haptoglobina – wiąże wolną Hb w surowicy. Kompleksy są niszczone w układzie siateczkowo-śródbłonkowym. W hemolizie wewnątrznaczyniowej może spaść do wartości niewykrywalnych.
5. Hiperbilirubinemia – bilirubina to produkt katabolizmu hemu, uwalnianego głównie z Hb w układzie siateczkowo-śródbłonkowym. Dlatego jest lepszym wskaźnikiem hemolizy zewnątrznaczyniowej. Duży wzrost stężenia bilirubiny może wskazywać na towarzyszącą dysfunkcję wątroby.
6. Ferrytyna – pośredni wskaźnik zasobów żelaza w organizmie. Jej stężenie wzrasta w hemolizie z powodu uwalniania żelaza z hemolizowanych erytrocytów. Nasilona lub wydłużona hemoliza oraz wyrównawcze pobudzenie erytropoezy mogą jednak skutkować niedoborem żelaza, w którym obserwuje się zmniejszenie stężenia ferrytyny.
7. Hemoglobinuria – występuje podczas nasilenia hemolizy wewnątrznaczyniowej. Hb pojawia się w moczu bezpośrednio po napadzie i jest od razu z niego usuwana. Napady występują często w nocy i od tego pochodzi nazwa choroby.
8. Hemosydenuria – hemosyderyna odpowiada za brązowy kolor moczu, pojawia się podczas znaczącej hemolizy wewnątrznaczyniowej. Utrzymuje się nawet przez kilka tygodni od napadu hemolizy.
Dla hemolizy występującej w NNH charakterystyczny jest ujemny wynik testu antyglobulinowego Coombsa, co różnicuje ją z autoimmunizacyjną niedokrwistością hemolityczną.
CYTOMETRIA PRZEPŁYWOWA
Od lat 90. XX wieku cytometria przepływowa jest uważana za metodę z wyboru i złoty standard w rozpoznawaniu i monitorowaniu NNH. 1 Obecnie analizy dokonuje się przy użyciu cytometrii wysokoczułej. Podczas badania oceniana jest ekspresja białek kotwiczonych na powierzchni komórek za pomocą GPI przy użyciu odpowiednich odczynników i przeciwciał monoklonalnych. Coraz częściej wykonuje się analizę przy użyciu odczynnika FLAER (fluorescent labelled aerolysin). FLAER to znakowana fluorochromem toksyna produkowana przez Aeromonas hydrophila. Ma zdolność do swoistego wiązania z białkami kotwicy GPI oraz do lizy komórek, z którymi się wiąże. Dlatego jest markerem, który z dużą dokładnością ocenia stopień niedoboru białek kotwicy GPI w komórkach. 1
Materiałem do analizy jest krew pełna, pobrana na dowolny antykoagulant. Analizy dokonuje się na komórkach co najmniej dwóch rodzajów, w tym obowiązkowo na granulocytach. W przeciwieństwie do erytrocytów czas życia granulocytów o fenotypie NNH nie ulega skróceniu. Co więcej, obecność defektu na erytrocytach może być maskowana przez wcześniejsze transfuzje koncentratu krwinek czerwonych. Dlatego liczba granulocytów dokładniej odzwierciedla wielkość klonu komórek NNH.
Cytometria przepływowa umożliwia nie tylko identyfikację defektywnych komórek, lecz również ocenę wielkości klonu we wszystkich liniach komórkowych. Ocenia się również stopień niedoboru GPI na komórkach i na tej podstawie dzieli erytrocyty na III typy:
III – z normalną ekspresją białek powierzchniowych wymagających kotwicy GPI,
III – z obniżoną ekspresją (o ok. 10 proc.), o zwiększonej wrażliwości na układ dopełniacza,
III – z brakiem ekspresji, bardzo wrażliwe na lizę.
Na przebieg kliniczny NNH wpływają zarówno wielkość klonu, jak i stopień niedoboru GPI w populacjach erytrocytów. Klon komórek z deficytem GPI ma tendencję do ekspansji wraz z czasem trwania choroby, dlatego zaleca się, aby cytometrię powtarzać co 3 miesiące.
ROZMAZ KRWI OBWODOWEJ I OBRAZ SZPIKU KOSTNEGO
Zmiany możliwe do stwierdzenia w rozmazie zarówno krwi obwodowej, jak i szpiku kostnego zależą od kategorii NNH.
W klasycznej NNH bez nieprawidłowości w szpiku kostnym w rozmazie krwi obwodowej obecna jest znaczna poikilocytoza (różnice w kształcie erytrocytów), anizocytoza (różnice w wybarwieniu) zwiększenie odsetka retikulocytów, mogą się pojawić erytroblasty. Szpik jest bogatokomórkowy, erytropoeza normoblastyczna, w wyniku wzmożonej wyrównawczej erytropoezy występuje hiperplazja układu czerwonokrwinkowego.
W NNH w przebiegu innych wad szpiku rozmaz krwi obwodowej oraz obraz szpiku wynikają z jego podstawowej choroby. W anemii aplastycznej charakterystyczny jest ubogokomórkowy obraz szpiku. Liczba komórek krwiotwórczych wszystkich linii jest zmniejszona, podwyższa się stosunek komórek tłuszczowych do komórek krwiotwórczych (>3). W rozmazie krwi widać zmniejszoną liczbę komórek wszystkich linii, o prawidłowym wyglądzie. Zmniejsza się odsetek retikulocytów. W przebiegu zespołów mielodysplastycznych szpik jest bogatokomórkowy, z nagromadzeniem nieprawidłowych, zmienionych morfologicznie komórek. Na obwodzie występuje cytopenia (jednej lub kilku linii komórkowych), w rozmazie można zauważyć anizocytozę, poikilocytozę, erytroblasty.
BADANIA GENETYCZNE
Znalezienie mutacji w genie PIG-A potwierdza diagnozę NNH. 1 Jednak z powodu trudności technicznych i finansowych badania genetyczne w kierunku mutacji PIG-A nie są wykonywane rutynowo.
CECHY KLINICZNE
Niedokrwistość
W momencie diagnozy anemia jest obecna u prawie wszystkich pacjentów. Natomiast masywna hemoliza wewnątrznaczyniowa prowadząca do hemoglobinurii dokumentowana jest jedynie u 25 proc. 11 Nazwa choroby ma znaczenie historyczne i pochodzi od napadów hemoglobinurii występujących w nocy, kiedy fizjologiczny spadek pH zwiększa działanie układu dopełniacza, powodując nasilenie hemolizy. Obecnie wiadomo, że hemoliza wewnątrznaczyniowa nie jest procesem napadowym, ale zachodzącym stale u osób chorych na NNH. Spadek pH oraz nasilenie hemolizy mogą być również wyzwalane przez intensywny wysiłek fizyczny, gorączkę, odwodnienie czy leki (diclofenak, ibuprofen, acetaminofen, penicylina, inhibitory β-laktamazy, erytromycyna, tetracykliny, rybawiryna, fludarabina, kladrybina, cisplatyna 1 ) oraz inne substancje chemiczne (benzen, ołów). Należy pamiętać, że niedokrwistość w przebiegu NNH może wynikać zarówno z hemolizy erytrocytów, jak i niewydolności szpiku. Na obecność hemolizy wskazują biochemiczne markery hemolizy. Za niewydolnością szpiku przemawiają m.in.:
- nieprawidłowości w morfologii krwi (leukopenia i trombocytopenia),
- spadek/brak wzrostu retikulocytów,
- prawidłowe stężenie LDH w surowicy.
Trombofilia
Epizody zakrzepowo-zatorowe to najczęstsza przyczyna śmierci w przebiegu NNH. 1 Dokumentowane są u 40 proc. pacjentów w momencie rozpoznania. Zakrzepica żylna występuje częściej niż tętnicza. Charakterystyczna jest nietypowa lokalizacja – najczęściej występuje jako zespół Budda-Chiariego (zakrzepica żył wątrobowych). 7 Inne częste lokalizacje to:
- żyła wrotna,
- żyła krezkowa,
- żyły mózgowe,
- żyły skórne.
Największe ryzyko zakrzepicy występuje u pacjentów z dużym odsetkiem komórek klonu NNH (>50 proc. granulocytów). 1 Charakterystyczna jest słaba odpowiedź na leczenie doustnymi antykoagulantami. Zakrzepica może współwystępować z trombocytopenią (w przebiegu niewydolności szpiku).
Na powstawanie predyspozycji do zakrzepicy może mieć wpływ wiele czynników. Postuluje się rolę dopełniacza, którego aktywacja powoduje uwalnianie prozapalnych i prozakrzepowych cytokin, takich jak IL- 6, IL-8, TNF-α. 1 Co więcej, niektóre białka (np. urokinase-type plasminogen activator receptor) biorące udział w procesie fibrynolizy również wymagają kotwicy GPI. 1 Ich brak/niedobór uzasadniałby częstsze epizody zakrzepowo-zatorowe. Wydaje się, że w dużym stopniu do trombofilii przyczynia się spadek stężenia tlenku azotu (NO), który ma właściwości antyagregacyjne. W procesie hemolizy wolna hemoglobina jest wiązana w surowicy przez haptoglobinę. Po wysyceniu wiązania hemoglobina przyczynia się do zmniejszenia stężenia NO, z czym związany jest efekt prozakrzepowy. 11
Dystonia mięśni gładkich (smooth muscle dystonia)
NO w warunkach fizjologicznych odpowiada nie tylko za efekt antyagregacyjny, lecz również za relaksację mięśni gładkich. Zatem jego spadek może się objawiać różnorodnymi zaburzeniami ze strony narządów, spowodowanymi zaburzoną relaksacją mięśni gładkich. Pacjenci z NNH mogą się skarżyć na:
- bóle brzucha (57 proc.),
- dysfagię (41 proc.),
- zaburzenia erekcji (47 proc.).
Dolegliwości nasilają się wraz ze zwiększeniem nasilenia hemolizy. 1
Nadciśnienie płucne
Pacjenci z NNH są narażeni na rozwój nadciśnienia płucnego z powodu skurczu naczyń w łożysku płucnym i zaburzenia relaksacji mięśni gładkich w wyniku zmniejszenia stężenia NO.
Przewlekła choroba nerek (PChN)
Pacjenci z NNH są obciążeni nawet sześciokrotnie większym ryzykiem przewlekłej choroby nerek. 1 Wynika to z destrukcji kanalików spowodowanej mikrozakrzepami oraz nadmiarem hemoglobiny uwolnionej z erytrocytów i złogami hemosyderyny odkładającymi się w kanalikach nerkowych.
U KOGO PODEJRZEWAC NNH?
PNH IG sugeruje, że na badania cytometryczne w kierunku NNH powinni być kierowani pacjenci, u których występuje:
- hemoglobinuria,
- dodatnie markery hemolizy wewnątrznaczyniowej, o ujemnym teście antyglobulinowym Coombsa, w szczególności z towarzyszącym niedoborem żelaza,
- choroba zakrzepowo-zatorowa, w szczególności o nietypowej lokalizacji (zespół Budda-Chiariego, żyły wrotne, krezkowe, mózgowe, skórne),
- anemia plastyczna,
- zespoły mielodysplastyczne z jedno- i wieloliniową dysplazją,
- dysfagia lub bóle brzucha z towarzyszącymi markerami hemolizy wewnątrznaczyniowej.
LECZENIE
Leczenie objawowe
Głównym celem objawowego leczenia jest zmniejszanie niedokrwistości spowodowanej hemolizą poprzez przetoczenia krwinek czerwonych, doustną suplementację żelaza oraz kwasu foliowego w przypadku stwierdzenia ich niedoboru (wyrównawcze nasilenie erytropoezy powoduje wzrost zapotrzebowania organizmu na te pierwiastki). W przypadku znacznie nasilonej niedokrwistości zaleca się podawanie koncentratów krwinek czerwonych (KKCz), należy jednak pamiętać, że wielokrotne i regularne transfuzje mogą się przyczynić do rozwinięcia wtórnej hemochromatozy. Terapia hormonami steroidowymi jest przedmiotem debat. Choć skuteczność długoterminowej terapii glikokortykosteroidami nie została potwierdzona, uważa się, że korzystne jest podawanie prednizonu u pacjentów z przełomem hemolitycznym.
Ostre epizody zakrzepowo-zatorowe leczone są fibrynolitycznie. Zasadność pierwotnej profilaktyki przeciwzakrzepowej jest dyskutowana. Dużym utrudnieniem jest mała skuteczność doustnych antykoagulantów oraz współwystępowanie małopłytkowości. Przed wdrożeniem leczenia należy wziąć pod uwagę wielkość klonu, wiek, choroby współistniejące, preferencje oraz pochodzenie pacjenta – obecne dane wskazują na częstsze występowanie zakrzepicy u mieszkańców Europy i Stanów Zjednoczonych.2] U pacjentów po przebytym zdarzeniu zakrzepowo-zatorowym wskazane jest dożywotnie leczenie przeciwzakrzepowe.
W przypadku NNH w przebiegu niewydolności szpiku leczenie powinno być ukierunkowane na niewydolność szpiku.
Ekulizumab
Ekulizumab to humanizowane przeciwciało monoklonalne, które blokuje działanie końcowych etapów aktywacji układu dopełniacza. W 2007 roku zostało zatwierdzone przez FDA do leczenia NNH. Dotąd pozostaje jedynym zarejestrowanym i używanym w praktyce klinicznej inhibitorem układu dopełniacza. 12
Ekulizumab przyłącza się do białka C5 i blokuje jego rozpad na C5a i C5b. W efekcie hamowane jest powstawanie MAC i skompensowany zostaje brak/niedobór CD59 na komórkach NNH, w związku z czym nie dochodzi do wewnątrznaczyniowej hemolizy erytrocytów. Przeciwciało to nie wpływa jednak na wcześniejsze etapy aktywacji dopełniacza. Brak/niedobór CD55 nie zostaje skompensowany, więc szlak konwertazy C3 prowadzący do opsonizacji erytrocytów przez C3b pozostaje nadmiernie aktywny. Dlatego u pacjentów leczonych ekulizumabem ujawniają się objawy toczącej się hemolizy zewnątrznaczyniowej. U ponad 50 proc. pacjentów następuje konwersja z ujemnego na dodatni test antyglobulinowy Coombsa. 1 25-35 proc. leczonych nadal wymaga transfuzji, a odpowiedź na ekulizumab dodatnio koreluje ze wzrostem klonu NNH (komórki są chronione przed lizą) oraz z poziomem hemolizy zewnątrznaczyniowej.
Ekulizumab wykazuje dużą skuteczność w leczeniu NNH. Badania kliniczne pokazały, że efektywnie blokuje hemolizę wewnątrznaczyniową, zmniejsza/znosi zapotrzebowanie na transfuzje, zmniejsza ryzyko zakrzepicy oraz podnosi jakość życia. 1, 2, 3, 4, 5 Opublikowane dotychczas wyniki dotyczące długoterminowej terapii przeciwciałem monoklonalnym pokazują, że również wydłuża czas przeżycia chorych (5-letnie przeżycie 96,5 proc. porównywane do historycznych wyników wynoszących ok. 65 proc. 1
Mimo skuteczności leku terapia ekulizumabem ma jednak pewne ograniczenia. Tworzenie MAC to kluczowy etap odpowiedzi odpornościowej oraz ochrony organizmu przed infekcjami. Największym ryzykiem związanym z blokadą układu dopełniacza jest podatność na zakażenia otoczkowymi bakteriami, w tym Neisseria meningitidis. 12 Dlatego wszyscy pacjenci otrzymujący lek powinni być zaszczepieni przeciwko wszystkim znanym szczepom N. meningitidis przed rozpoczęciem leczenia.
Główną przeszkodą dla dostępności leczenia jest koszt ekulizumabu. Chociaż kryteria kwalifikacji do leczenia nie zostały jednoznacznie określone, powinno być ono zarezerwowane dla najcięższych przypadków choroby. Co więcej, ekulizumab nie wpływa na funkcję szpiku, dlatego pacjenci z NNH w przebiegu lub z towarzyszącą niewydolnością szpiku odnoszą najmniejsze korzyści z leczenia. 7
Z odpowiedzią na ekulizumab związany jest polimorfizm genu CR1. Jego produkt, białko błonowe CR1, jest kofaktorem dla czynnika I rozkładającego C4b i C3b. Jak wspomniano, u chorych na NNH jest jedynym sprawnie działającym błonowym regulatorem dopełniacza w komórkach o fenotypie NNH. Polimorfizm HindiIII odpowiada za niską ilość CR1 na powierzchni komórek (genotyp L/L) oraz siedmiokrotnie większe ryzyko słabej (sub-optimal) odpowiedzi na leczenie. 1 Co więcej, w 2014 roku u Japończyków zidentyfikowano punktową mutację zmiany sensu, c.2654G-àA, która powoduje zmianę C5 w miejscu przyłączenia przeciwciała i zapobiega przyłączaniu się leku. 1 Mutacja występuje w 3,2 proc. populacji japońskiej i nie została dotychczas wykryta u mieszkańców innych krajów.
W związku z ograniczeniami ekulizumabu (wysokie koszty leczenia, ryzyko infekcji meningokokowej, niedostateczna odpowiedź u niektórych pacjentów, objawy hemolizy zewnątrznaczyniowej) nowe cząsteczki są przedmiotami badań. Nowe badane inhibitory dopełniacza, takie jak mini FH czy białka fuzyjne FH-CR2, blokują układ dopełniacza na wcześniejszych etapach, głównie na poziomie C3, i wykorzystują CR2 i czynnik F (naturalny rozpuszczalny regulator UD). 1
Przeszczepienie komórek krwiotwórczych
Jedyną metodą dającą możliwość całkowitego wyleczenia NNH jest przeszczepienie alogenicznych komórek krwiotwórczych. Obarczone jest ono jednak trudnością w postaci doboru dawcy, wysokim ryzykiem powikłań oraz śmiertelnością – przede wszystkim z powodu infekcji oraz ostrej i przewleklej choroby przeszczep przeciwko gospodarzowi. 1, 2, 3 Dlatego PNH IG zaznacza, że powinno być rozważane tylko w wyjątkowych przypadkach: współistnienia niewydolności szpiku (PNH/AA, PNH/MDS), nawracającej zagrażającej życiu choroby zakrzepowo zatorowej opornej na leczenie przeciwzakrzepowe lub w opornej na leczenie ekulizumabem postaci niedokrwistości hemolitycznej zależnej od przetoczeń KKCz. Po allo-SCT od dawców w pełni zgodnych w układzie HLA (10/10) dwuletnie przeżycie wynosi 56 proc., a 10-letni wskaźnik przeżycia 42 proc. Przeciętna częstość występowania choroby przeszczep przeciwko gospodarzowi to 40-50 proc. Natomiast chorobę zamykania żył wątrobowych VOD (veno-occlusive disease of liver) obserwowano u połowy chorych. 38 U części chorych obserwowano powikłania zakrzepowo-zatorowe. Analiza retrospektywna przeprowadzona przez grupę francuską wykazała, że przeżycie całkowite pacjentów z NNH po allo-SCT powikłanym zakrzepicą jest znacząco niższe niż w grupie bez zakrzepicy. 36 Przeszczepienie KKM i jego powikłania wpływają również na pogorszenie jakości życia pacjentów. 39, 40 . Dlatego allo-SCT nie jest leczeniem pierwszego wyboru u chorych z klasyczną postacią NNH.
U pacjentów, u których zaplanowano allo-SCT, ekulizumab był stosowany rzadko i jest niewiele doniesień na ten temat, a grupy opisanych chorych tak leczonych są nieliczne. Leczenie ekulizumabem może być stosowane do kontroli hemolizy jednocześnie z przeszczepem KKM. W przypadku rzutów hemolizy po allo-SCT zalecana jest dodatkowa dawka leku. 41, 42
Ze względu na brak kompleksowych badań decyzję o ewentualnej kwalifikacji chorego do allo-SCT należy indywidualizować, biorąc pod uwagę przebieg kliniczny i czynniki ryzyka mogące doprowadzić do śmierci. Po wykonaniu allo-SCT badanie klonu NNH należy wykonywać co trzy miesiące aż do momentu, gdy klon komórek NNH nie będzie wykrywany, a następnie badanie to wykonujemy raz w roku. 43
Abstract
ABSTRACT
Paroxysmal nocturnal hemoglobinuria (PNH) is a rare hemolytic anemia caused by a non-malignant clonal expansion of hematopoietic stem cells with a somatic mutation in the PIG-A gene. PIG-A has its locus on the X chromosome and its products are involved in early steps of glycosylphosphatidylinositol (GPI) anchor synthesis in the cell membrane. The resulting defect in GPI anchor synthesis in hematopoietic stem cells leads to a deficiency/lack of the surface proteins CD55 and CD59 responsible for the inhibition of spontaneous complement activation, and therefore to excessive and uncontrolled activity of the complement system. The clinical presentation involves intravascular hemolysis, bone marrow failure and thrombosis. A negative antiglobulin Coombs test with hemolytic markers is a distinctive laboratory finding. Thromboembolic episodes often occur at atypical sites (with Budd-Chiari syndrome in the first place) and are the most common cause of death in the course of the disease. The patients are at higher risk of developing pulmonary hypertension, chronic kidney disease and smooth muscle dystonia (dysphagia, abdominal pain, erectile dysfunction). Flow cytometry is a gold standard of diagnosing and monitoring PNH, and should be repeated every 3 months. Symptomatic treatment includes red blood cell transfusions and oral supplementation of iron and folic acid (in the case of their deficiency). Prednisone is said to be beneficial in patients in hemolytic crisis. Eculizumab is a monoclonal antibody registered for PNH treatment. By binding to the C5 protein, it inhibits late steps of complement activation and thus compensates for the deficiency/lack of CD59 on the cell surface. It effectively inhibits intravascular hemolysis, reduces/eliminates the need for transfusions and decreases the risk of thrombosis, improving the quality of life and overall survival. The only curative method of treating PNH is allogeneic stem cell transplantation. Due to a high risk of complications and mortality, the decision should be individualized, considering the clinical course of the disease and the presence of potentially life-threatening risk factors.
KEYWORDS: paroxysmal nocturnal hemoglobinuria, hemolytic anemia, clonal expansion, hematopoietic stem, PIG-A gene mutation. cells transfusions.
- 1. Rosse WF, Nishimura J. Clinical manifestations of paroxysmal nocturnal hemoglobinuria: present state and future problems. Int J Hematol 2003;77:113-20
- 2. Parker C, Omine M, Richards S, Nishimura J, Bessler M, Ware R, Hillmen P, Luzzatto L, Young N, Kinoshita T, Rosse W, Socié G. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood, 2005;106:3699-709
- 3. Żupańska B, Spychalska J, Pyl H, Mendek-Czajkowska E, Brojer E. Nocna napadowa hemoglobinuria – wieloletnie obserwacje. Charakterystyka kliniczna i analiza wielkości klonu z defektem kotwicy glikozylofosfatydyloinozytolowej (GPI). Acta Haematologica Polonica;43(1):75-82
- 4. Gołąd J, Jakóbisiak M, Lasek W, Stokłosa T. Immunologia. Wydawnictwo Naukowe PWN,2014;211
- 5. Harboe M, Mollnes TE. The alternative complement pathway revisited. J. Cell. Mol. Med. Vol 12, No 4, 2008;1074-84
- 6. Risitano AM, Notaro R, Marando L et al. Complement fraction 3 binding on erythrocytes as additional mechanism of disease in paroxysmal nocturnal hemoglobinuria patients treated by eculizumab. Blood. 2009;113(17):4094-100
- 7. Brodsky RA. Paroxysmal nocturnal hemoglobinuria. Blood. 2014;124:2804-11
- 8. Kummar V, Abbas AK, Aster JC. Robbins Patologia, Wroclaw, Elsevier 2014;457
- 9. Luzzatto L. Paroxysmal nocturnal hemoglobinuria: an acquired X-linked genetic disease with somatic-cell mosaicism. Current Opinion in Genetics & Development 2006;16:317-22
- 10. Hu R, Mukhina GL, Piantadosi S, Barber JP, Jones RJ, Brodsky RA: PIG-A mutations in normal hematopoiesis. Blood 2005;105:3848-54
- 11. Schubert J, Reoth A. Update on paroxysmal nocturnal haemoglobinuria: on the long way to understand the principles of the disease. European Journal of Haematology 94;464-73
- 12. Edwin K, Wong S, Kavanagh D. Anticomplement C5 therapy with eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome. Translational Research, 2015
- 13. Barcellini W, Fattizzo B. Clinical Applications of Hemolytic Markers in the Differential Diagnosis and Management of Hemolytic Anemia. Disease Markers 2015; 635-70
- 14. Wróbel A. Nocna napadowa hemoglobinuria w świetle najnowszych badań. Hematologia 2011;(2):4:346-8
- 15. Brodsky RA, Mukhina GL, Li S, Nelson KL, Chiurazzi PL, Buckley JT, Borowitz MJ. Improved detection and characterization of paroxysmal nocturnal hemoglobinuria using fluorescent aerolysin. Am J Clin Pathol. 2000 Sep; 114(3):459-66
- 16. Luzzatto L, Nafa K. Genetics of PNH. In: Young NS, Moss J, eds. Paroxysmal Nocturnal Hemoglobinuria and the Glycosylphosphatidylinositol-Linked Proteins. San Diego, CA: Academic Press; 2000;21-47
- 17. Hałka J, Sułek K. Choroby krwi wywołane przez leki. Acta Haematologica Polonica 2010;41:2:131-9
- 18. Hill A, Kelly RJ, Hillmen P. Thrombosis in paroxysmal nocturnal hemoglobinuria. Blood. 2013;121(25):4985-96
- 19. Moyo VM, Mukhina GL, Garrett ES, Brodsky RA. Natural history of paroxysmal nocturnal haemoglobinuria using modern diagnostic assays. Br J Haematol. 2004;126(1):133-8
- 20. Ritis K, Doumas M, Mastellos D et al. A novel C5a receptor-tissue factor cross-talk in neutrophils links innate immunity to coagulation pathways. J Immunol. 2006;177(7):4794-802
- 21. Maroney SA, Cunningham AC, Ferrel J et al. A GPI-anchored co-receptor for tissue factor pathway inhibitor controls its intracellular trafficking and cell surface expression. J Thromb Haemost. 2006;4(5):1114-24
- 22. Parker CJ, Ware RE. Paroxysmal nocturnal hemoglobinuria w: Greer JP, Foerster J, Lukens JN, Rodgers GM, Paraskevas F, Glader BE. eds. Wintrobe’s Clinical Hematology. 11th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2004;1203-21
- 23. Hillmen P, Elebute M, Kelly R et al. Long-term effect of the complement inhibitor eculizumab on kidney function in patients with paroxysmal nocturnal hemoglobinuria. Am J Hematol. 2010;85(8):553-9
- 24. Luzzatto L, Gianfaldoni G, Notaro R. Management of Paroxys¬mal nocturnal haemoglobinuria: a personal view. Br. J. Haematol. 2011;153:709-20
- 25. Hill A, Rother RP, Arnold L et al. Eculizumab prevents intravascular hemolysis in patients with paroxysmal nocturnal hemoglobinuria and unmasks low level extravascular hemolysis occurring through C3 opsonization. Haematologica. 2010;95(4):567-73
- 26. Hillmen P, Muus P, Dührsen U, Risitano AM, Schubert J, Luzzatto L, Schrezenmeier H, Szer J, Brodsky RA, Hill A, Socié G, Bessler M, Rollins SA, Bell L, Rother RP, Young NS. Effect of the complement inhibitor eculizumab on thromboembolism in patients with paroxysmal nocturnal hemoglobinuria. Blood. 2007;110(12):4123-8
- 27. Kelly RJ, Hill A, Arnold LM, Brooksbank GL, Richards SJ, Cullen M, Mitchell LD, Cohen DR, Gregory WM, Hillmen P. Long-term treatment with eculizumab in paroxysmal nocturnal hemoglobinuria: sustained efficacy and improved survival. Blood. 2011;117(25):6786-92
- 28. Risitano AM. Paroxysmal Nocturnal Hemoglobinuria in the era of complement inhibition. ‘Accepted Article’, doi: 10.1002/ajh.24323
- 29. Hillmen P, Muus P, Roth A et al. Long-term safety and efficacy of sustained eculizumab treatment in patients with paroxysmal nocturnal haemoglobinuria. Br J Haematol 2013;162:62-73
- 30. Rother RP, Rollins SA, Mojcik CF, Brodsky RA, Bell L. Discovery and development of the complement inhibitor ekulizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat Biotechnol 2007;25:1256-64
- 31. Hillmen P, Muus P, Roth A et al. Long-term safety and efficacy of sustained eculizumab treatment in patients with paroxysmal nocturnal haemoglobinuria. Br J Haematol 2013;162:62-73
- 32. Rondelli T, Risitano AM, Peffault de Latour R, Sica M, Peruzzi B, Ricci P, Barcellini W, Iori AP, Boschetti C, Valle V, Fremeaux-Bacchi V, De Angioletti M, Socie G, Luzatto L, Notaro R. Polymorphism of the complement receptor 1 gene correlates with the hematologic response to eculizumab in patients with paroxysmal nocturnal hemoglobinuria. Haematoogica 2014;99(2)
- 33. Nishimura J, Yamamoto N, Hayashi S, Ohyashiki K, AndoAndres K, Brodsky RA., Noji H, Kitamura K, Eto T, Takahashi T, Masuko M, Matsumoto T, Wano Y, Shichishima T, Shibayama H, Hase M, Li L, Johnson K, Lazarowski , Tamburini P, Inazawa J, Kinoshita T, Kanakura Y. Genetic Variants in C5 and Poor Response to Eculizumab. N Engl J Med 2014;370:632-9
- 34. Schmidt CQ, Hardera MJ, Nichols E, Hebeckerc M, Anlikerd M, Höchsmannd B, Simmet T, Csincsie AI, Uzonyif B, Pappworthb IY, Rickling D, Lambrisg JD, Schrezenmeierd H, Józsie M, Marchbank KJ. Selectivity of C3-opsonin targeted complement inhibitors: A distinct advantage in the protection of erythrocytes from paroxysmal nocturnal hemoglobinuria patients. Immunobiology 221, 2016;503-11
- 35. Saso R, Marsh J, Cevreska L et al. Bone marrow transplants for paroxysmal nocturnal haemoglobinuria.Br J Haematol. 1999;104:392-6
- 36. Peffault de Latour R, Schrezenmeier H, Bacigalupo A et al. Allogeneic stem cell transplantation in paroxysmal nocturnal hemoglobinuria. Haematologica. 2012;97(11): 1666-73
- 37. Santarone S, Bacigalupo A, Risitano AM Tagliaferri E, Di Bartolomeo E, Iori AP, Rambaldi A, Angelucci E, Spagnoli A, Papineschi F, Tamiazzo S, Di Nicola M, Di Bartolomeo P. Hematopoietic stem cell transplantation for paroxysmal nocturnal hemoglobinuria: long-term results of a retrospective study on behalf of the Gruppo Italiano Trapianto Midollo Osseo (GITMO). Haematologica 2010;95:983-8
- 38. Fraser CJ, Bhatia S, Ness K Francisco L, Arora M, Parker P, Forman S, Weisdorf D, Gurney JG, Baker KS. Impact of chronic graft-versus-host disease on the health status of hematopoietic cell transplantation survivors: a report from the Bone Marrow Transplant Survivor Study. Blood 2006;108:2867-73
- 39. Bieri S, Roosnek E, Helg C Verholen F, Robert D, Chapuis B, Passweg J, Miralbell R, Chalandon Y. Quality of life and social integra¬tion after allogeneic hematopoietic SCT. Bone Marrow Transplant 2008;42:819-27
- 40. Goker H, Uz B, Buyukasik Y, Aksu S, Haznedaroglu I, Sayınalp N, Karacan Y, Tekin F, Ozcebe OI. Eculizumab before and after alloge¬neic hematopoietic stem cell transplantation in a patient with paroxysmal nocturnal hemo¬globinuria: Case report. Turk J Hematol 2011;28:223-7
- 41. Taniguchi K, Okada M, Yoshihara S, Sawada A, Tokugawa T, Ishii S, Kaida K, Ikegame K, Minagawa K, Matsui T, Ogawa H. Strategy for bone marrow transplantation in eculizumab-treated paroxysmal nocturnal hemoglobinuria. Int J Hematol 2011;94:403-7
- 42. Sahin F, Saydam G, Pesg PNH diagnosis, follow-up and treatment guidelines Am J Blood Res 2016;6(2):19-27
Następny artykuł:
Nocna napadowa hemoglobinuria – od teorii do praktyki klinicznej


 Dodaj do ulubionych
Dodaj do ulubionych
