


Co znajdziesz w artykule?
- W artykule omówiono problem hepatopatii polekowych w trakcie leczenia onkologicznego. Opisano ich mechanizmy, a także zebrano najczęściej indukujące je leki, łącznie z omówieniem tych, które są aktualnie dostępne w terapii polekowego uszkodzenia wątroby (DILI – drug-induced liver injury)
Spis treści
- Mechanizmy polekowego uszkodzenia wątroby w praktyce klinicznej
- Diagnostyka polekowego uszkodzenia wątroby
- Objawy polekowego uszkodzenia wątroby
- Hepatopatie występujące w konsekwencji leczenia choroby nowotworowej
- Farmakoterapia u kresu życia a ryzyko hepatotoksyczności leków
- Farmakoterapia hepatopatii polekowych
- Czego należy unikać w leczeniu, by nie doszło do hepatopatii polekowych
Polekowe uszkodzenia wątroby coraz częściej stają się przyczyną zaburzeń czynności tego organu, choć postawienie właściwego rozpoznania nie jest łatwe. Obraz kliniczny tego typu zaburzeń jest różnorodny i waha się od niewielkiego, często przemijającego podwyższenia aktywności enzymów indykatorowych wątroby do jej ostrej niewydolności. Pacjenci onkologiczni należą do szczególnych grup ryzyka uszkodzenia wątroby, co wynika z jednej strony z choroby nowotworowej, z drugiej zaś ze stosowanego
leczenia. Nie do pominięcia jest, że sami chorzy i ich rodziny często poszukują alternatywnych terapii, co wielokrotnie doprowadza do wystąpienia hepatopatii polekowych. Warto przypomnieć, że w leczeniu systemowym plasują się one na 3 miejscu pod względem częstotliwości występowania uszkodzeń wątroby bezpośrednio po paracetamolu i lekach stosowanych u pacjentów z wirusem upośledzenia odporności (HIV – human immunodeficiency virus). Objawy DILI, które występują w praktyce po jednym leku, są rzadkie i dotyczą reakcji idiosynkratycznych, natomiast w praktyce najczęściej dochodzi do hepatopatii będących konsekwencją interakcji o typie farmakokinetycznym i sumowania działań niepożądanych poszczególnych farmaceutyków, leków roślinnych i suplementów diety 1, 2, 3 . Warto przypomnieć, że interakcje leków i ich konsekwencje kliniczne stają się coraz większym problemem praktycznie we wszystkich specjalnościach lekarskich. Powszechna polifarmakoterapia sprawia, że pacjenci przyjmują coraz więcej preparatów, które wchodząc między sobą w interakcje farmakokinetyczne i farmakodynamiczne, indukują objawy chorób polekowych, spośród których jedną z klinicznie istotniejszych są właśnie hepatopatie polekowe. Problem ten komplikuje również skłonność chorych z nowotworem do samoleczenia oraz suplementowania, co sprawia, że analiza przyczyn eskalacji aktywności enzymów indykatorowych staje się trudna, choć nie niemożliwa. Warto pamiętać, że 1, 3 :
- objawy hepatopatii polekowych może indukować aż 900 różnych preparatów
- 20-40% przypadków niewydolności wątroby jest powodowanych przez leki
- 75% idiosynkratycznych reakcji wątrobowych jest pochodzenia polekowego
- 2-5% pacjentów hospitalizowanych z powodu żółtaczki ma cechy DILI.
Do istotnych klinicznie czynników ryzyka wystąpienia polekowego uszkodzenia wątroby zaliczane są 1, 3 :
- wcześniej występujące choroby wątroby
- płeć – u kobiet częściej w porównaniu z mężczyznami występują polekowe objawy niepożądane związane z tym organem
- wiek – im starszy pacjent, tym ryzyko wystąpienia hepatotoksyczności jest większe
- czynniki genetyczne
- alkoholizm
- polifarmakoterapia, zwłaszcza z udziałem leków intensywnie metabolizowanych przy udziale cytochromu P450 (CYP450).
U pacjenta onkologicznego istnieją bardziej złożone mechanizmy oraz czynniki ryzyka prowadzące do uszkodzenia wątroby. Są nimi:
- preparaty stosowane w leczeniu onkologicznym
- leki wykorzystywane w chorobach współistniejących, które mogą wykazywać działanie hepatotoksyczne
- suplementy diety o działaniu hepatotoksycznym lub które wpływają na zwiększenie ryzyka hepatotoksyczności stosowanych jednocześnie farmaceutyków
- uszkodzenia wątroby indukowane radioterapią (RTH – radiotherapy)
- żywienie pozajelitowe
- współistniejące choroby wątroby
- nowotwory złośliwe pierwotne i wtórne wątroby
- powikłania zakrzepowe (żyły wrotna i wątrobowe)
- zespoły paraneoplastyczne.
Nie należy również zapominać o czynnikach, które mogą predysponować do hepatopatii polekowych. Najistotniejszymi z nich są 3, 4, 5 :
- dawka leku
- profil metaboliczny
- ryzyko wystąpienia interakcji między lekami
- polifarmakoterapia
- stosowanie suplementów diety
- używanie roślinnych produktów leczniczych
- przyjmowanie witamin, zwłaszcza w wysokich dawkach
- wiek
- płeć
- otyłość
- wielochorobowość
- profil farmakogenetyczny
- palenie papierosów
- spożywanie alkoholu.
Z praktyki wynika, że leki będące substratami enzymów CYP450 mają większe prawdopodobieństwo wywołania DILI w sposób niezależny od dawki, podczas gdy te będące inhibitorami CYP450 tylko wówczas, gdy są podawane w wysokich dziennych dawkach. Ekspresja CYP450 jest zależna od polimorfizmów genetycznych, rodzaju ksenobiotyków, cytokin i hormonów 3, 4, 5 .
Mechanizmy polekowego uszkodzenia wątroby w praktyce klinicznej
Wśród patomechanizmów DILI wyróżnienia się, takie jak 1, 4, 5, 6 :
- indukcja stresu oksydacyjnego w wątrobie (spożywanie alkoholu, zbyt duża podaż żelaza) powodująca generację dużej ilości wolnych rodników, które aktywują komórki Kupffera
- dysfunkcja mitochondriów
- zakłócenie procesu transkrypcji
- tworzenie adduktów (powstawanie wiązań kowalencyjnych), co powoduje utratę funkcji i/lub reakcje autoimmunologiczne
- zahamowanie transportu wątrobowego
- indukcja/inhibicja enzymów metabolizujących leki
- reakcje idiosynkratyczne.
W tabeli 1 zebrano farmaceutyki często stosowane w praktyce klinicznej, które sprzyjają wystąpieniu hepatopatii polekowych 3, 6 .
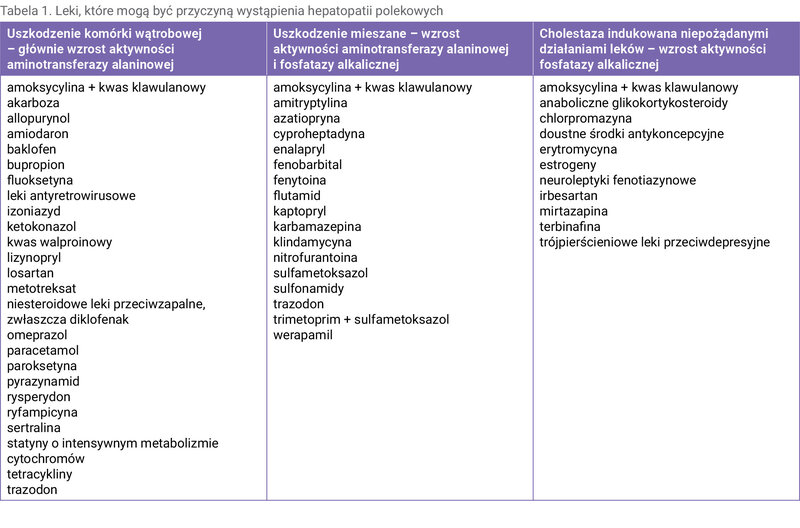
Tabela 1. Leki, które mogą być przyczyną wystąpienia hepatopatii polekowych
Diagnostyka polekowego uszkodzenia wątroby
Problemem diagnostyki DILI jest to, że stosowane markery nie są specyficzne względem tego zaburzenia wątroby. Wzrost aktywności aminotransferazy alaninowej (AlAT – alanine aminotransferase), która jest głównym sygnałem do zdiagnozowania DILI, jest również obserwowany w wielu innych schorzeniach, w tym w chorobach nowotworowych oraz w przypadku wirusowego zapalenia czy stłuszczenia wątroby. Z kolei zwiększone stężenie bilirubiny występuje również w nadczynności tarczycy i chorobach kości. Za tzw. złoty standard w przewidywaniu poważnego DILI uważa się prawo Hya. Zakłada ono porównanie aktywności AlAT (3-krotnie przekroczona norma) i stężenia bilirubiny w surowicy (2-krotnie powyżej normy) przy jednocześnie występującej żółtaczce. Współcześnie potrzebne są jednak modyfikacje tej zależności, które pozwalałyby szybciej i skuteczniej oceniać ryzyko uszkodzeń wątroby na skutek stosowania leków. Do tego mogą przyczynić się nowe biomarkery DILI 4, 5, 6 .
Objawy polekowego uszkodzenia wątroby
Do najczęstszych objawów klinicznych toksycznego uszkodzenia wątroby zalicza się:
- nadmierną męczliwość
- jadłowstręt
- nudności
- ogólne osłabienie
- utratę apetytu
- powiększenie wątroby i śledziony
- świąd skóry.
Należy pamiętać, że znaczna część objawów opisywanych przez pacjenta onkologicznego może wynikać zarówno z samej choroby, jak i działań niepożądanych stosowanej terapii. Rokowanie u chorych z DILI jest na ogół korzystne, ponieważ u większości dochodzi do regresji zmian klinicznych i laboratoryjnych po odstawieniu hepatotoksycznego leku. Niemniej u tych z miąższowym uszkodzeniem wątroby ponad 3-krotnie przekroczona norma stężenia bilirubiny (>3 mg/dl) jest związana z 10% ryzykiem zgonu. W takich przypadkach należy natychmiast odstawić podejrzewany o interakcje farmaceutyk. W przypadkach DILI bez żółtaczki i ze wzrostem AlAT mniejszym niż 3-krotność normy można tylko zmniejszyć dawkę leku, kontynuując monitorowanie aktywności transaminaz. U niektórych pacjentów w wyniku zjawiska adaptacji dochodzi do wycofania się nieprawidłowości laboratoryjnych bez odstawienia leku 4, 5, 6 .
Hepatopatie występujące w konsekwencji leczenia choroby nowotworowej
Tabela 2 zawiera leki stosowane w chemioterapii (CTH – chemotherapy) pod względem ryzyka wystąpienia DILI 3 .
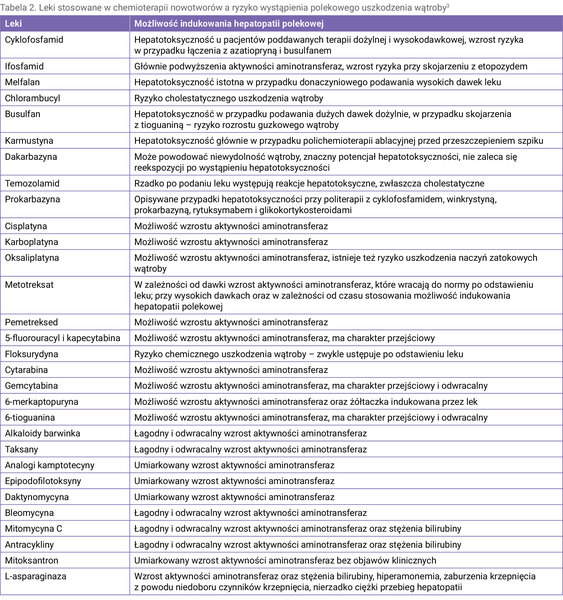
Tabela 2. Leki stosowane w chemioterapii nowotworów a ryzyko wystąpienia polekowego uszkodzenia wątroby3
Ryzyko hepatotoksyczności związane jest również ze stosowaniem hormonoterapii, immunoterapii, a także leków ukierunkowanych molekularnie oraz inhibitorów kinaz białkowych. W hormonoterapii takimi farmaceutyki są 3 :
- tamoksyfen
- fulwestrant
- octan cyproteronu
- flutamid
- bikalutamid
- nilutamid
- enzalutamid
- abirateron.
Niski potencjał hepatotoksyczności związany jest ze stosowaniem inhibitorów aromatazy, a także analogów gonadoliberyny 3 .
Przy immunoterapii i lekach ukierunkowanych molekularnie należy pamiętać o ryzyku hepatotoksyczności, zwłaszcza w przypadku stosowania interfenonu α (IFN-α), interleukiny 2 (IL-2), ipilimumabu oraz rytuksymabu. Niskie ryzyko w rozumieniu farmakoterapii kontekstowej związane jest z leczeniem bewacyzumabem, cetuksymabem, panitumumabem oraz trastuzumabem 3 .
Inhibitorami kinaz białkowych, których stosowanie zwiększa ryzyko występowania hepatopatii polekowych, są:
- afatynib
- erlotynib
- gefitynib
- imatynib
- lapatynib
- pazopanib
- regorafenib
- sorafenib
- sunitynib
- wemurafenib.
Niskie ryzyko wystąpienia hepatotoksyczności jest związane ze stosowaniem aksytynibu oraz dabrafenibu.
Oczywiście ryzyko hepatotoksyczności u chorego z nowotworem nie może być rozpatrywane wyłącznie przez pryzmat pojedynczego preparatu. Należy pamiętać o szczególnych interakcjach lek–choroba, które w istotny sposób modyfikują bezpieczeństwo farmakoterapii w tej szczególnej populacji pacjentów. W tabeli 3 zebrano istotne cechy leczenia farmaceutycznego, które w praktyce ma na celu redukcję ryzyka występowania działań niepożądanych, zwłaszcza hepatotoksyczności 1, 2, 3 .

Tabela 3. Czynniki zależne od farmakoterapii wpływające na ograniczenie ryzyka wystąpienia hepatotoksyczności
Farmakoterapia u kresu życia a ryzyko hepatotoksyczności leków
Szczególną grupą są pacjenci w terminalnym okresie chorób nowotworowych, u których kontynuowane jest wg wskazań klinicznych stosowanie różnych grup leków onkologicznych. W tym szczególnym okresie wzrasta ryzyko wystąpienia DILI związanych z farmakoterapią (tab. 4).
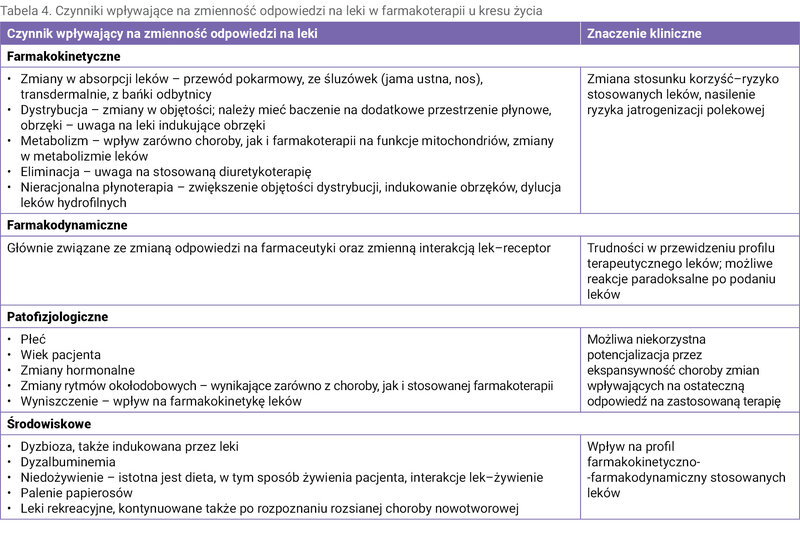
Tabela 4. Czynniki wpływające na zmienność odpowiedzi na leki w farmakoterapii u kresu życia
Jak już wspomniano, chorzy z nowotworem często poszukują terapii alternatywnych, leków roślinnych i suplementów diety. Mogą one niestety powodować reakcje hepatotoksyczne, które często przyczyniają się do zaprzestania leczenia z uwagi na przeciwwskazania poszczególnych preparatów. Składnikami roślinnych produktów leczniczych i suplementów diety, które mogą indukować zaburzenia funkcji wątroby, są 3 :
- tamaryndowiec malabarski
- zielona herbata
- pluskwica groniasta
- czosnek
- pieprz metystynowy
- jeżówka purpurowa
- lakownica żółta
- ostryż długi
- witania ospała (ashwagandha).
Należy także ograniczyć stosowanie produktów leczniczych i suplementów diety zawierających witaminę A.
Farmakoterapia hepatopatii polekowych
L-asparaginian L-ornityny
L-asparaginian L-ornityny (LOLA – L-ornithine L-aspartate) bierze udział w dwóch najważniejszych procesach detoksykacji amoniaku powstającego w wyniku przemian białek – syntezie mocznika oraz syntezie glutaminy. Jest aktywatorem enzymów biorących udział w tych przemianach oraz substratem do syntezy mocznika. Wskazaniami do stosowania LOLA są encefalopatia wątrobowa (EW) oraz pomocnicze leczenie zaburzeń czynności wątroby różnego pochodzenia. Lek ma praktyczne znaczenie z uwagi na mechanizm działania w przypadku hepatopatii polekowych. Przy niewydolności wątroby jako konsekwencji niepożądanego działania farmaceutyków LOLA staje się lekiem z wyboru 7, 8, 9 .
Nie zaleca się stosowania suplementów diety zawierających LOLA, m.in. z powodu zawartości zbyt niskiego stężenia asparaginianu ornityny, co może powodować ich nieskuteczność, a nawet działania niepożądane. L-asparaginian L-ornityny jest solą dwóch aminokwasów endogennych: ornityny i kwasu asparaginowego. Zarówno L-ornityna, jak i L-asparaginian wchodzą w skład cyklu mocznikowego (cykl Krebsa) w wątrobie i są niezbędne w procesie syntezy mocznika i usuwania amoniaku z organizmu. L-asparaginian uczestniczy ponadto w wiązaniu amoniaku z kwasem glutaminowym do glutaminy w mięśniach szkieletowych. W ten sposób podaż LOLA stymuluje oba procesy wiążące amoniak w organizmie oraz przeciwdziała hiperamonemii w stanach niewydolności wątroby i wzmożonego katabolizmu tkankowego. Lek jest dobrze tolerowany i bezpieczny nawet u pacjentów z ciężką niewydolnością wątroby. Z uwagi na mechanizm działania i wieloczynnikowość patogenezy EW w marskości wątroby LOLA powinno się stosować u chorych z hiperamonemią 7, 9, 10 . Istotne jest dawkowanie:
- dożylne we wlewie: z szybkością maksymalnie do 5 g/godzinę, zwykle do 20 g/24 godziny, w stanach przedśpiączkowych i hepatopatiach polekowych oraz w śpiączce – do 40 g/24 godziny
- doustnie: 1-2 saszetki granulatu 1-3 ×/24 godziny; lek należy przyjmować w trakcie posiłku lub po nim.
Kwas tiazolidynokarboksylowy
Kwas tiazolidynokarboksylowy (TCA) jest metabolizowany w wątrobie do cysteiny, która dzięki obecności grupy tiolowej bierze udział w procesach detoksykacji etanolu, a także leków. Łatwo wchłania się z przewodu pokarmowego, przenika w postaci niezmienionej do wątroby. Tam ulega przemianie do N-formylocysteiny, która w swej cząsteczce zawiera wolną grupę −SH, niezbędną do powstawania wielu połączeń tiolowych, biorących udział w przemianach wątrobowych, m.in. w procesie detoksykacji. Lek stosuje się pomocniczo w ostrych i przewlekłych chorobach wątroby o różnej etiologii oraz w toksycznym uszkodzeniu wątroby spowodowanym alkoholem, lekami lub substancjami chemicznymi. Powoduje szybką normalizację stężenia enzymów indykatorowych wątroby. Początkowe dawkowanie to 200 mg 2-3 ×/24 godziny, a następnie, po ustąpieniu ostrych objawów – 100 mg 2 ×/24 godziny.
Kwas ursodeoksycholowy
Kwas ursodeoksycholowy (UDCA) jest silnie hydrofilnym i prawie nietoksycznym kwasem żółciowym. Ma wiele właściwości, które odróżniają go od innych kwasów żółciowych. Wykazuje m.in. silne działanie żółciopędne, cytoprotekcyjne, szczególnie na hepatocyty oraz cholangiocyty, ma właściwości antyapoptotyczne i immunomodulacyjne. Od wielu lat UDCA stosowany jest w różnych chorobach wątroby i dróg żółciowych, zwłaszcza tych, które przebiegają z cholestazą, w hepatopatiach polekowych.
Z długiej listy wszystkich potencjalnych efektów farmakologicznych UDCA najlepiej udokumentowano trzy mechanizmy i na nich opierają się prawie wszystkie aktualne wskazania do stosowania UDCA w terapii. Są nimi:
- ochrona cholangiocytów przed cytotoksycznym działaniem hydrofobowych kwasów żółciowych
- pobudzanie wydzielania żółci w wątrobie
- ochrona hepatocytów przed apoptozą stymulowaną przez inne kwasy żółciowe.
W praktyce klinicznej UDCA stosuje się w szerokim zakresie w dawkach 10-40 mg/kg m.c./24 godziny w 2-3 dawkach, niemniej optymalna wydaje się dawka 15 mg/kg m.c./24 godziny.
Fosfolipidy
Fosfolipidy wprowadzone do organizmu wbudowują się w błony komórkowe oraz uszkodzone hepatocyty. Fosfatydylocholina poprawia czynność układów enzymatycznych oraz receptorów błonowych. Lek zwiększa zdolności regeneracyjne hepatocytów, a także stabilizuje i ochrania ich błony komórkowe przed działaniem czynników toksycznych. Preparaty zawierające fosfolipidy zalecane są w:
- ostrych i przewlekłych chorobach wątroby
- uszkodzeniach wątroby (toksycznych, polekowych, poalkoholowych), najlepiej w skojarzeniu z produktami leczniczymi, które wspomagają procesy detoksykacyjne w wątrobie
- wspomagająco w leczeniu zaburzeń lipidowych wątroby
- leczeniu chorób dróg żółciowych i kamicy żółciowej
- pomocniczo w chorobach wirusowych wątroby.
Brakuje jednoznacznych danych, na podstawie których można zalecać fosfolipidy w DILI. Jeśli podejmuje się takie próby, to terapia powinna być skojarzona z lekami o innych mechanizmach działania hepatoprotekcyjnego.
Czego należy unikać w leczeniu, by nie doszło do hepatopatii polekowych
Należy unikać suplementów diety, a także produktów zawierających ostropest plamisty. Wyciągi z niego hamują aktywność izoenzymów CYP450: CYP3A4, CYP2D6, CYP2C9, indukując interakcje z wieloma lekami, czego konsekwencją mogą być DILI, w tym także u pacjentów onkologicznych. Brakuje badań uzasadniających stosowanie sylimaryny w polekowych uszkodzeniach wątroby 11, 12 . Dużą ostrożność należy zachować podczas stosowania produktów o działaniu żółciotwórczym i żółciopędnym.
Abstract
ABSTRACT
Drug-induced liver damage during cancer treatment. Practical tips to remember
Drug-induced liver damage is a common cause of liver dysfunction, although this diagnosis is not easy to establish. Even though it is becoming more common, it often goes undiagnosed. The clinical presentation of drug-induced liver damage is diverse, ranging from a slight, often transient increase in the activity of liver indicator enzymes to acute liver failure. Cancer patients are at increased risk of liver damage associated both with the cancer and with the treatment administered.
- 1. Hansten PD, Horn JR. The Top 100 drug interactions. H&H Publications, Dunedin 2019
- 2. Bazire S. Psychotropic drug directory. Lloyd-Reinhold Publications, London 2021
- 3. Shear NH. Drug eruption & reaction manual. CRC Press, Boca Raton 2023
- 4. Ghabril M, Chalasani N, Bjornsson E. Drug-induced liver injury: a clinical update. Curr Opin Gastroenterol 2010;26(3):222-6
- 5. Padda MS, Sanchez M, Akhtar AJ, et al. Drug-induced cholestasis. Hepatology 2011;53(4):1377-87
- 6. Rusyn I, Bataller R. Alcohol and toxicity. J Hepatol 2013;59(2):387-8
- 7. Mato JM, Lu SC. Role of S-adenosyl-L-methionine in liver health and injury. Hepatology 2007;45(5):1306-12
- 8. Mato JM, Camara J, de Paz JF, et al. S-adenosylmethionine in alcoholic liver cirrhosis: a randomized, placebo-controlled, double-blind, multicenter clinical trial. J Hepatol 1999;30(6):1081-9
- 9. Feld JJ, Modi AA, El-Diwany R, et al. S-adenosyl methionine improves early viral responses and interferon-stimulated gene induction in hepatitis C nonresponders. Gastroenterology 2011;140(3):830-9
- 10. Papakostas GI, Mischoulon D, Shyu I, et al. S-adenosyl methionine (SAMe) augmentation of serotonin reuptake inhibitors for antidepressant nonresponders with major depressive disorder: a double-blind, randomized clinical trial. Am J Psychiatry 2010;167(8):942-8
- 11. Kvasnicka F, Biba B, Sevcik R, et al. Analysis of the active components of silymarin. J Chromatogr A 2003;990(1-2):239-45
- 12. Loguercio C, Festi D. Silybin and the liver: from basic research to clinical practice. World J Gastroenterol 2011;17(18):2288-301

 Dodaj do ulubionych
Dodaj do ulubionych
