



Co znajdziesz w artykule?
- Klasyfikacja, objawy kliniczne oraz ustalenie typu i podtypu choroby von Willebranda
- Ocena aktywności i stężenia vWF oraz aktywności FVIII podstawą diagnostyki choroby von Willebranda
- Zastosowanie desmopresyny oraz koncentratu FVIII zawierającego vWF (FVIII/vWF) w leczeniu choroby von Willebranda
Spis treści
Choroba von Willebranda jest najczęściej rozpoznawaną wrodzoną skazą krwotoczną (1:1000 osób w populacji). Do najczęstszych jej objawów należą: krwawienia z nosa, wzmożone siniaczenie się, obfite, przedłużone krwawienia miesiączkowe oraz krwawienia po ekstrakcjach zębów i zabiegach operacyjnych. Wyróżniamy 3 podstawowe typy choroby von Willebranda:
- typ 1 – najczęściej rozpoznawany (75%), z proporcjonalnym zmniejszeniem stężenia i aktywności czynnika von Willebranda (vWF – von Willebrand
- factor) oraz z łagodnym nasileniem krwawień
- typ 2 – występują w nim zaburzenia różnych funkcji vWF, a nasilenie krwawień jest zwykle umiarkowane
- typ 3 – charakteryzuje się ciężkimi krwawieniami oraz nieoznaczalnym stężeniem i aktywnością vWF (3-5%).
Historia
W 1926 roku fiński lekarz Erik von Willebrand opisał nową jednostkę chorobową. Zaobserwował u członków rodzin zamieszkujących Wyspy Alandzkie krwawienia prowadzące nawet do śmierci w młodym wieku, np. wskutek obfitych, przedłużonych krwawień miesiączkowych. Choroba występowała zarówno u płci męskiej, jak i żeńskiej 1 . W badaniach prowadzonych w drugiej połowie XX wieku wyodrębniono w 1971 roku białko o budowie multimerycznej, którego niedobór lub nieprawidłowa budowa są odpowiedzialne za obraz tej choroby, nazwanej od nazwiska odkrywcy chorobą von Willebranda. Białko to nazwano czynnikiem von Willebranda. W 1985 roku cztery niezależne grupy badaczy sklonowały komplementarne DNA vWF, a w 1991 roku budowę genu vWF przedstawili Sadler i wsp. 2 .
Czynnik von Willebranda
Czynnik vW to duża i złożona glikoproteina osoczowa odgrywająca ważną rolę w procesie hemostazy. Produkowany jest on w komórkach śródbłonka i megakariocytach. Początkowo jest syntezowany w postaci pojedynczego łańcucha o masie 300 kDa, który ulega dimeryzacji i multimeryzacji, tworząc wielkocząsteczkowe multimery wydzielane do osocza. Czynnik vW magazynowany jest w ciałkach Weibla-Palade’a komórek śródbłonka. Odgrywa istotną rolę w pierwotnej hemostazie, umożliwiając adhezję płytek do ściany naczynia w miejscu jego uszkodzenia oraz agregację płytek krwi. Ponadto chroni czynnik VIII (FVIII – factor VIII) przed proteolityczną degradacją poprzez wiązanie go z domeną znajdującą się w N-końcowym segmencie vWF, co zapewnia mu stabilność w osoczu. Gen vWF znajduje się na krótkim ramieniu chromosomu 12 i zawiera 178 kb (kilo par zasad).
Epidemiologia
Choroba von Willebranda jest diagnozowana w populacji z częstością 1:100 osób w badaniach przesiewowych, natomiast jej objawowe postaci występują 10-krotnie rzadziej 3 . Ze względu na często skąpoobjawowy przebieg choroba von Willebranda może być rozpoznana dopiero u dorosłych. Według danych Instytutu Hematologii i Transfuzjologii w Polsce zarejestrowanych jest około 2000 chorych. Wobec tego ogromna większość przypadków jest niezgłoszona i nierozpoznana 4 .
Objawy kliniczne
Do najczęstszych objawów występujących u osób z rozpoznaną chorobą von Willebranda należą: nadmierne siniaczenie się, krwawienia z nosa i błon śluzowych jamy ustnej, przedłużone, obfite krwawienia miesiączkowe, krwawienia po ekstrakcji zębów i zabiegach operacyjnych. Rzadko, tylko w ciężkiej postaci choroby, dochodzi do krwawień śródstawowych, w wyniku których może wystąpić artropatia hemofilowa, oraz jeszcze rzadziej do krwawień śródczaszkowych. Krwawienia z przewodu pokarmowego związane z angiodysplazją u pacjentów z typem 3, typem 2A i 2B choroby von Willebranda są bardziej charakterystyczne dla dorosłych.
Klasyfikacja choroby von Willebranda
Choroba von Willebranda cechuje się dużą heterogennością. Oprócz trzech podstawowych typów – 1 i 3 charakteryzujących się ilościowym zmniejszeniem vWF i typu 2 związanego z jakościowym zaburzeniem funkcji tego czynnika – w typie 2 wyróżnia się różne podtypy.
Typ 1 to najczęstsza postać, stanowiąca około 75% wszystkich przypadków choroby von Willebranda. Występujące u pacjentów krwawienia są zwykle łagodne lub umiarkowanie ciężkie. Aktywność vWF i antygen vWF są proporcjonalnie zmniejszone, a aktywność FVIII może być prawidłowa lub zmniejszona. Czas częściowej tromboplastyny po aktywacji (APTT – activated partial thromboplastin time) może być prawidłowy. Autorzy międzynarodowych wytycznych zalecają zakwalifikować pacjentów ze znacznym obniżeniem antygenu vWF (<30%n) i fenotypem krwotocznym do typu 1, natomiast chorych z pośrednim obniżeniem vWF (30-50%n) zaliczyć do oddzielnej kategorii nazwanej niskim vWF 5, 6 .
W typie 2 choroby von Willebranda wyróżniono wiele podtypów. Objawy klinicznie mają zwykle charakter umiarkowanych krwawień. Zaburzenia dotyczą różnych fizjologicznych funkcji vWF. Rozróżnienie typu 1 i typu 2 choroby opiera się głównie na określeniu stosunku aktywności vWF, wyrażonej jako aktywność kofaktora rystocetyny vWF (vWF:RCo [ristocetin cofactor]) do antygenu vWF (vWF:Ag). W typie 2 stosunek ten wynosi ≤0,6.
W podtypie 2A – najczęstszym wariancie typu 2 – występuje zmniejszona adhezja płytek zależna od vWF z niedoborem wielkocząsteczkowych multimerów. Aktywność vWF jest znacznie obniżona, podczas gdy antygen i FVIII mogą być prawidłowe lub zmniejszone w niewielkim stopniu. Dziedziczenie jest autosomalne dominujące lub recesywne.
W podtypie 2B stwierdza się zwiększone powinowactwo vWF do płytkowej glikoproteiny Ib (GPIb). Skutkuje to usuwaniem wielkocząsteczkowych multimerów vWF z krążenia i w wyniku tego dochodzi do krwawień. Zwykle u chorych występuje małopłytkowość nasilająca się pod wpływem stresu, zabiegów operacyjnych, infekcji. Wyniki badań laboratoryjnych pacjentów z podtypem 2B są podobne do uzyskiwanych u chorych z wariantem 2A, ale charakterystyczna dla podtypu 2B jest zwiększona agregacja płytek krwi pod wpływem małego stężenia rystocetyny (LD-RIPA – low-dose ristocetin-iduced platelet aggregation). Dziedziczenie jest autosomalne dominujące.
W podtypie 2M choroby von Willebranda występuje prawidłowa liczba wielkocząsteczkowych multimerów vWF, ale adhezja płytek krwi zależna od vWF jest osłabiona wskutek zaburzonej reakcji między vWF, GPIb płytek oraz elementami tkanki łącznej. Dziedziczy się autosomalnie dominująco lub recesywnie.
W podtypie 2N dochodzi do zmniejszonej zdolności wiązania FVIII przez vWF. W wyniku tego przy prawidłowym vWF aktywność FVIII jest zmniejszona, zwykle do wartości <10%n. Test wiązania FVIII przez vWF pozwala na różnicowanie podtypu 2N z łagodną hemofilią A, ponadto jego dziedziczenie jest autosomalne.
Przyczyną choroby von Willebranda typu płytkowego jest defekt płytkowej glikoproteiny Ibα (GPIbα). Powoduje on nasilenie wiązania płytek do vWF, co prowadzi do małopłytkowości i utraty wielkocząsteczkowych multimerów vWF. Krwawienia są zwykle łagodne lub umiarkowane. U części pacjentów rozpoznawany jest podtyp 2B choroby. Dziedziczy się autosomalnie dominująco.
Pacjenci z typem 2 choroby von Willebranda, których nie udało się zaklasyfikować do żadnego podtypu, określani są mianem nieustalonych (2NS – not stated).
U osób z rozpoznaną chorobą von Willebranda typu 3 krwawienia występują już w okresie niemowlęcym. Pacjenci łatwo, nadmiernie się siniaczą, pojawiają się u nich krwawienia z nosa, błon śluzowych jamy ustnej, miesiączki są bardzo obfite, długie i prowadzą do ciężkiej niedokrwistości, występują krwawienia do stawów i mięśni. Przyczyną jest brak vWF, co powoduje bardzo małą aktywność FVIII. Dziedziczenie jest autosomalne recesywne.
W tabeli 1 przedstawiono charakterystykę typów choroby von Willebranda.
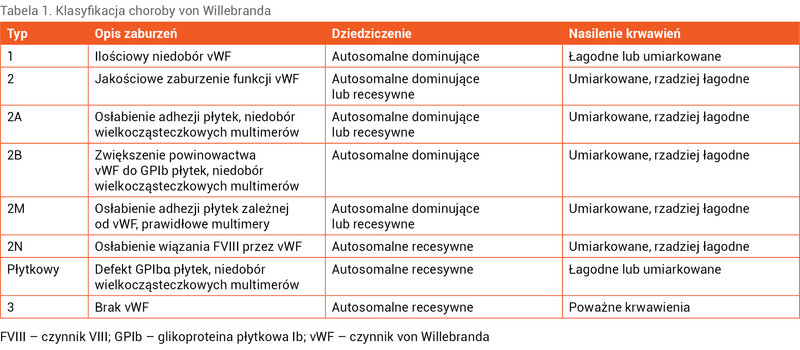
Tabela 1. Klasyfikacja choroby von Willebranda
Ocena krwawień w diagnostyce choroby von Willebranda jest bardzo ważna. W tym celu stosuje się wystandaryzowaną skalę oceny krwawień u dzieci i dorosłych, która była modyfikowana kilka razy. Obecnie zaleca się opracowaną przez International Society on Thrombosis and Haemostasis skalę Bleeding Assessment Tool (ISTH-BAT), według której ocenia się jakościowo (lokalizacja i przyczyna krwawienia, np. miesiączka, błona śluzowa nosa, jamy ustnej, staw, ekstrakcja zęba, zabieg operacyjny; w sumie 14 punktów) oraz ilościowo (nasilenie krwawienia; 0-4 punktów) historię krwawień u pacjenta. Maksymalnie można osiągnąć 56 punktów. Prawidłowy wynik u dziecka to <3, a u kobiety 0-5 punktów 7 .
Badania diagnostyczne w chorobie von Willebranda
Chorobę von Willebranda rozpoznaje się na podstawie wywiadu dotyczącego krwawień, wywiadu rodzinnego oraz badań laboratoryjnych.
Do podstawowych badań laboratoryjnych w kierunku tej choroby należą: aktywność vWF mierzona jako kofaktor rystocetyny (vWF:RCo), stężenie vWF – antygen (vWF:Ag) oraz aktywność FVIII.
APTT, którego wydłużenie związane jest ze zmniejszoną aktywnością FVIII, może być prawidłowy, ponieważ często w typach 1 i 2 choroby von Willebranda FVIII jest w granicach normy. Czas okluzji badany w aparacie PFA-100 jest wydłużony u większości pacjentów, ale nie jest to badanie zalecane jako przesiewowe w kierunku choroby von Willebranda. Trzeba pamiętać, że wiele czynników ma wpływ na wyżej wymienione wyniki. Stres, wysiłek, infekcja, stany zapalne, nowotwory, zabiegi operacyjne, doustne środki antykoncepcyjne, ciąża, a nawet miesiączka zwiększają stężenia vWF i FVIII. Z tego względu należy w miarę możliwości wyeliminować te czynniki lub wziąć je pod uwagę przy interpretacji wyników badań. Po uzyskaniu obniżonych wartości powinno się powtórzyć badania w celu potwierdzenia rozpoznania, zwłaszcza gdy objawy krwotoczne są małe lub nie ma ich wcale. Z drugiej strony niewielkie objawy krwotoczne mogą występować również u dzieci zdrowych. U dzieci do 6 miesiąca życia typ 1 choroby von Willebranda nie powinien być wykluczany ze względu na podwyższone stężenia vWF w okresie noworodkowym i wczesnym niemowlęcym 8 .
Jeśli obniżenie vWF:RCo i vWF:Ag nie jest proporcjonalne, należy obliczyć stosunek tych parametrów w celu różnicowania typu 1 i 2. Współczynnik vWF:RCo/vWF:Ag <0,6 wskazuje na typ 2 choroby von Willebranda. W dalszej diagnostyce typu 2 wskazane jest badanie multimerów vWF. W podtypach 2A, 2B i płytkowym wielkocząsteczkowe multimery są obniżone. W celu ustalenia, czy pacjent ma podtyp 2A, czy 2B, wykonuje się badanie LD-RIPA. Wynik jest dodatni w podtypie 2B (≥30%). Aby odróżnić podtyp 2B od płytkowego (rzekomego), można przeprowadzić test wiązania vWF badanej osoby z prawidłowymi płytkami pod wpływem rystocetyny (RIPA – ristocetin induced platelet agglutination). W przypadku płytkowego podtypu choroby von Willebranda wiązanie jest prawidłowe.
Wyniki badań laboratoryjnych w typach i podtypach choroby von Willebranda przedstawiono w tabeli 2.
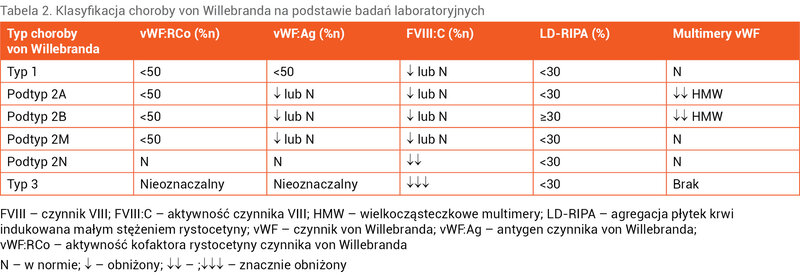
Tabela 2. Klasyfikacja choroby von Willebranda na podstawie badań laboratoryjnych
Badania genetyczne
Badania genetyczne mogą być pomocne w rozpoznaniu choroby von Willebranda, a także różnicowaniu jej typów i podtypów. W typie 1 z vWF <30%n u większości pacjentów wykrywane są zmiany w genie vWF (91,8%). Natomiast przy tzw. niskim poziomie vWF (30-50%) wykrywalność zaburzeń w genie jest w granicach 40-64% pacjentów 5, 9 . Badania genetyczne mogą być wykorzystane w diagnostyce prenatalnej typu 3 choroby, zwłaszcza gdy rodzice mają już chore dziecko. W tym celu przeprowadza się analizy DNA kosmków kosmówki. Można również badać mutację sprawczą choroby wśród członków rodziny z wykrywaniem nosicielstwa w przypadku typu choroby z recesywnym dziedziczeniem. W różnicowaniu łagodnej postaci hemofilii i podtypu 2N choroby von Willebranda, w przypadku braku wywiadu rodzinnego, badanie genetyczne może być kluczowe, gdy nie można wykonać testu wiązania FVIII z vWF 8 .
Leczenie pacjentów z chorobą von Willebranda
Podstawowym lekiem stosowanym w chorobie von Willebranda jest desmopresyna (DDAVP – 1-deamino-8-D-argininowazopresyna), która jest syntetyczną pochodną wazopresyny – hormonu antydiuretycznego. Desmopresyna uwalnia vWF zmagazynowany w komórkach śródbłonka, powodując 2-5-krotne zwiększenie aktywności vWF i FVIII w osoczu. Lek podawany jest dożylnie w dawce 0,3 µg/kg m.c. w 50-100 ml 0,9% NaCl w kroplówce trwającej 30 minut. Maksymalny wzrost aktywności vWF i FVIII następuje w ciągu 30-90 minut od zastosowania leku. Desmopresynę można podawać donosowo w dawce 150 µg przy masie ciała do 50 kg i 300 µg przy masie ciała większej niż 50 kg. W grupie dziewcząt z przedłużającymi się, obfitymi krwawieniami miesiączkowymi, które odpowiadają na leczenie desmopresyną, donosowa forma leku jest szeroko stosowana.
Lek ten można stosować u większości pacjentów z typem 1 choroby von Willebranda, ponieważ wykazują oni odpowiedni wzrost vWF i FVIII. W typie 2 skuteczność desmopresyny może być mała, a w podtypie 2B może dojść do przejściowej małopłytkowości. W przypadku podtypu 2N czas półtrwania FVIII może być znacznie skrócony. W typie 3 choroby lek ten nie działa 10 . Wśród działań niepożądanych desmopresyny wymienia się: ból głowy, uderzenia gorąca, zmiany ciśnienia krwi, hiponatremię z zatrzymaniem wody, co może wywołać drgawki 11 . Nie zaleca się podawania leku dzieciom poniżej 2 roku życia. Przed zastosowaniem leku zaleca się przeprowadzenie u chorych testu z desmopresyną. Polega on na oznaczeniu aktywności vWF i FVIII przed podaniem leku oraz po godzinie od jego zastosowania. W przypadku podejrzenia zwiększonego klirensu leku (gorszej skuteczności) wskazane jest kolejne oznaczenie po 4 godzinach. Po 3-4 dniach słabnie efekt podawania DDAVP z powodu wyczerpania rezerw tkankowych czynników krzepnięcia, wobec tego przy poważnych krwawieniach lub podczas zabiegów operacyjnych konieczne jest zastąpienie desmopresyny koncentratem FVIII zawierającym vWF (FVIII/vWF).
Koncentrat FVIII/vWF jest lekiem z wyboru w leczeniu: pacjentów z typem 3 choroby von Willebranda, chorych z typem 2, gdy desmopresyna jest nieskuteczna, a także pacjentów z typem 1 choroby, którzy wymagają dłuższej terapii. Ze względu na pierwotny niedobór vWF zalecane są preparaty zawierające więcej vWF niż FVIII, o stosunku vWF:FVIII >1. Najbardziej polecany jest koncentrat, w którym stosunek vWF:FVIII wynosi 2,4 (Haemate P) 12, 13 . Inny koncentrat (Fanhdi) ma 1,2 raza więcej vWF niż FVIII. Koncentraty należy dawkować w oparciu o jednostki vWF/kg m.c. Wstrzyknięcie 1 j. vWF/kg m.c. powoduje wzrost vWF o około 2 j./dl (%n). Czas leczenia uzależniony jest od wielkości i lokalizacji krwawienia oraz od rodzaju zabiegu operacyjnego. Dostępne są również osoczopochodny koncentrat vWF praktycznie niezawierający FVIII (Wilfactin) oraz czysty rekombinowany vWF 13 . Preparatów tych nie stosowano dotąd w Polsce, ponadto rekombinowany vWF obecnie jest zarejestrowany tylko do leczenia dorosłych ze względu na brak odpowiednich badań w grupie dzieci.
Większość dzieci leczonych jest w razie wystąpienia krwawienia lub zabiegu operacyjnego, czyli sporadycznie. U wybranych pacjentów z typem 3 choroby von Willebranda stosuje się profilaktycznie długoterminowo koncentrat vWF/FVIII lub vWF, aby zapobiec krwawieniom do stawów lub częstym, nawracającym krwawieniom z nosa bądź z przewodu pokarmowego.
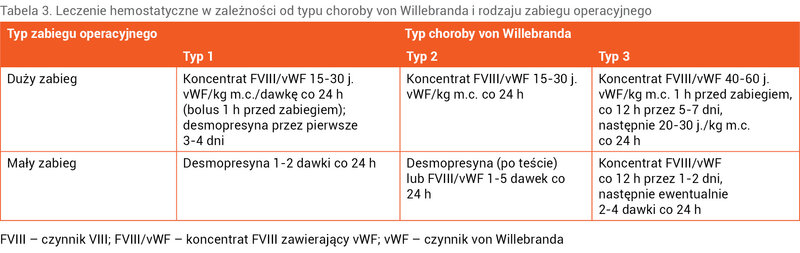
Tabela 3. Leczenie hemostatyczne w zależności od typu choroby von Willebranda i rodzaju zabiegu operacyjnego
W tabeli 3 przedstawiono dawkowanie leków podczas zabiegów operacyjnych.
Leczenie wspomagające
W chorobie von Willebranda bardzo istotną rolę odgrywają leki antyfibrynolityczne. Kwas traneksamowy jest skuteczny u dziewcząt z przedłużającymi się obfitymi krwawieniami miesiączkowymi, w przypadku krwawień z nosa i błon śluzowych jamy ustnej oraz podczas zabiegów przeprowadzanych w obrębie błon śluzowych jamy ustnej i nosa. Może być stosowany razem z desmopresyną lub koncentratem vWF/FVIII; jest on wystarczający w monoterapii u pacjentów z łagodnym przebiegiem postaci choroby i w sytuacji małych zabiegów lub krwawień. Zwykle stosuje się go przez 5-7 dni w dawce 20 mg/kg m.c. (maksymalnie 1,0 g) 3 razy dziennie. Może być podawany doustnie w postaci tabletek (0,5 g) i ampułek do picia (1 g/10 ml) lub w dożylnej kroplówce. Wśród obserwowanych objawów niepożądanych działania kwasu traneksamowego wymienia się: zawroty głowy, nudności, wymioty, biegunki oraz reakcje alergiczne.
W leczeniu krwawień z nosa skuteczna jest tamponada przednia z użyciem spongostanu nasączonego trombiną wołową. Spongostan stosowany jest również na zębodół po ekstrakcji zęba. Kleje fibrynowe wykorzystywane są podczas niektórych zabiegów chirurgicznych.
W przypadku trudnych do leczenia obfitych krwawień miesiączkowych skuteczne są doustne środki antykoncepcyjne 3 .
Ryzyko krwawień u dziewcząt z chorobą von Willebranda maleje w czasie ciąży ze względu na zwiększenie vWF. Nie dotyczy to 3 typu choroby. Badania aktywności vWF i FVIII powinno się wykonać w III trymestrze ciąży, aby ustalić postępowanie w okresie okołoporodowym. W okresie poporodowym wzrasta ryzyko wystąpienia krwawienia u pacjentek z chorobą von Willebranda ze względu na powrót vWF i FVIII do wartości wyjściowych, który następuje w ciągu 7-21 dni od urodzenia dziecka. Dziewczynę w ciąży należy objąć opieką specjalistyczną i ustalić plan leczenia hemostatycznego w razie wystąpienia krwawienia lub postępowania profilaktycznego.
Dziecko z typem 3 choroby von Willebranda powinno być szczepione podskórnie zamiast domięśniowo, natomiast nie jest to konieczne w pozostałych typach choroby.
Każde dziecko po rozpoznaniu choroby von Willebranda powinno otrzymać kartę chorego na wrodzoną skazę krwotoczną oraz kartę postępowania zawierającą informacje o leczeniu, aktualizowaną co 1-3 lata.
Koncentraty czynnika vW/VIII oraz desmopresynę pacjent lub zamawiający lekarz otrzymują bezpłatnie w Regionalnym Centrum Krwiodawstwa i Krwiolecznictwa po zgłoszeniu zapotrzebowania poprzez stronę Narodowego Centrum Krwi (https://csm-swd.nfz.gov.pl/cnr) lub po wpisaniu w wyszukiwarkę Google „czynnik na ratunek”.
Podsumowanie
Dostępne różne możliwości leczenia choroby von Willebranda pozwalają, podobnie jak w hemofilii, na personalizację terapii w zależności od problemów medycznych, oczekiwań oraz stylu życia pacjenta. Nowe wytyczne dotyczące diagnostyki i leczenia choroby von Willebranda, konsultowane przez ekspertów z American Society of Hematology, International Society on Thrombosis and Haemostasis, National Hemophilia Foundation i World Federation of Hemophilia, powstaną prawdopodobnie do końca 2020 roku 14 .
Abstract
Von Willebrand disease in children
Von Willebrand disease is the most common congenital bleeding disorder, although its mild course in most cases is the reason why the disease is not so often recognised as haemophilia. Inheritance is autosomal dominant or recessive. Typical symptoms of disease include mucosal bleedings from the nose and gums, easy bruising, heavy menstrual bleedings, and haemorrhages after tooth extraction and surgical events. There are 3 types of disease. Types 1 and 3 are characterised by a quantitative von Willebrand factor deficiency and type 2 by qualitative defects of structure and function of vWF. Type 1 is diagnosed in 75% of all patients whereas type 3, severe, with virtually complete deficiency of vWF, is found in 3–5% of patients. Diagnosis is based on clinical presentation, family history and laboratory test results. Activity of factor VIII and activity and antigen of von Willebrand factor are primary tests for diagnosing the disease. In differential diagnosis of some types and subtypes of vWD, some other tests are necessary, including multimer analysis, FVIII and FvW binding and platelet aggregation by low dose of ristocetin (LD-RIPA). Desmopressin is the first choice in most patients with type 1 vWD. In cases when the drug is not effective, as in type 3 vWD, or efficacy is low, FVIII/vW concentrates are used. Most patients are treated sporadically, in the case of bleeding or before a surgical procedure. Only in severe type 3 disease is continuous or prolonged prophylaxis recommended, mostly to stop joint bleeds.
- 1. Nilsson IM. The history of von Willebrand disease. Haemophilia 1999;5(Suppl 2):7-11
- 2. Ginsburg D. The molecular biology of von Willebrand disease. Haemophilia 1999;5(Suppl 2):19-27
- 3. Zdziarska J, Chojnowski K, Klukowska A. i wsp. Postępowanie w chorobie von Willebranda. Med Prakt 2008 (wydanie specjalne 12):3-23
- 4. Narodowy program leczenia chorych na hemofilię i pokrewne skazy krwotoczne na lata 2019-2023. Program Polityki Zdrowotnej. Ministerstwo Zdrowia, 2018
- 5. Lavin M, O'Donnell JS. How I treat low von Willebrand factor levels. Blood 2019;133(8):795-804
- 6. Castaman G, Goodeve A, Eikenboom J; European Group on von Willebrand Disease. Principles of care for the diagnosis and treatment of von Willebrand disease. Haematologica 2013; 98(5):667-74
- 7. Elbatarny M, Mollah S, Grabell J, et al. Normal range of bleeding scores for the ISTH-BAT: adult and pediatric data from the merging project. Haemophilia 2014;20(6):831-5
- 8. Laffan MA, Lester W, O'Donnell JS, et al. The diagnosis and management of von Willebrand disease: a United Kingdom Haemophilia Centre Doctors Organization guideline approved by the British Committee for Standards in Haematology. Br J Haematol 2014;167(4):453-65
- 9. Peake IR, Goodeve AC. Genetic testing for von Willebrand disease: the case for. J Thromb Haemost 2010;8(1):13-6
- 10. Windyga J, Dolan G, Altisent C, et al. Practical aspects of DDAVP use in patients with von Willebrand disease undergoing invasive procedures: a European survey. Haemophilia 2016;22(1):110-20
- 11. Stoof SC, Cnossen MH, de Maat MP, et al. Side effects of desmopressin in patients with bleeding disorders. Haemophilia 2016;22(1):39-45
- 12. de Jager NCB, Bukkems LH, Heijdra JM, et al. One piece of the puzzle: population pharmacokinetics of FVIII during perioperative Haemate P®/Humate P® treatment in von Willebrand disease patients. J Thromb Haemost 2020;18(2):295-305
- 13. Mannucci PM. New therapies for von Willebrand disease. Blood Adv 2019;3(21):3481-7
- 14. Kalot MA, Al-Khatib M, Connell NT, et al. An international survey to inform priorities for new guidelines on von Willebrand disease. Haemophilia 2020;26(1):106-16
Następny artykuł:

 Dodaj do ulubionych
Dodaj do ulubionych
