


Spis treści
Hemochromatoza wrodzona jest dziedziczną chorobą prowadzącą do nadmiernego gromadzenia się żelaza i w konsekwencji uszkodzenia wielu tkanek i narządów. Jest to najczęściej występujące schorzenie genetyczne rasy kaukaskiej, a badania nad nim przyczyniły się do głębszego poznania procesów związanych z gospodarką żelazową.
CELE ARTYKUŁU
Po przeczytaniu artykułu Czytelnik powinien znać:
• podstawowe informacje na temat patofizjologii hemochromatozy wrodzonej
• objawy hemochromatozy wrodzonej
•
podstawy diagnostyki i leczenia hemochromatozy wrodzonej
Nadmierne gromadzenie się żelaza w organizmie człowieka wynika z licznych zaburzeń o charakterze zarówno wrodzonym, jak i nabytym. Spośród nich hemochromatoza wrodzona, związana z mutacjami genu HFE (HFE-HC – HFE-hemochromatosis), jest najczęściej występującym (ale wciąż rzadko rozpoznawanym) schorzeniem genetycznym, dziedziczonym autosomalnie recesywnie. Większość chorych na hemochromatozę wrodzoną to nosiciele mutacji C282Y genu HFE, u których bez włączenia leczenia nadmiar żelaza może prowadzić do nieodwracalnego uszkodzenia narządów miąższowych oraz wynikającej z tego chorobowości i śmiertelności.
Gospodarka żelazowa
Żelazo jest pierwiastkiem niezbędnym w organizmie ludzkim m.in. do syntezy hemoglobiny, mioglobiny oraz katalizacji licznych procesów enzymatycznych. Dzienne wchłanianie wynosi ok. 1-2 mg, a ustrojowe zapasy tego pierwiastka sięgają 3-5 g. U chorych z hemochromatozą wrodzoną może dochodzić do dziennego wchłaniania nawet 10 mg żelaza, a zapasy ustrojowe wynoszą 20-40, a nawet 60 g. Nadmiar żelaza jest szkodliwy, gdyż nasila stres oksydacyjny w komórkach, a w wątrobie powoduje uszkodzenie mitochondriów, mikrosomów i lizosomów, czego skutkiem jest włóknienie i karcynogeneza. Najważniejszym hormonem regulującym gospodarkę żelazową u człowieka jest hepcydyna, produkowana głównie w wątrobie. Hepcydyna krążąca we krwi łączy się z obecną w makrofagach i enterocytach ferroportyną, co hamuje uwalnianie żelaza z tych dwóch rodzajów komórek. 1 Ekspresję hepcydyny zwiększają zarówno nadmiar żelaza, jak i stan zapalny w organizmie, a skutkuje to zmniejszonym wchłanianiem jelitowym żelaza oraz jego obniżonym uwalnianiem z makrofagów. Niedobór żelaza, a także nieefektywna erytropoeza czy niedotlenienie zmniejszają z kolei aktywność hepcydyny. 2 Na funkcję hepcydyny wpływają także wrodzone mutacje jej genu oraz innych genów biorących udział w gospodarce żelazowej, takich jak ferroportyna, HFE, hemojuwelina czy receptor transferyny typu 2, oraz czynniki nabyte – zakażenia wirusami hepatotropowymi, nadmierne spożywanie alkoholu czy stłuszczeniowa choroba wątroby. 3
Klasyfikacja zespołów przeładowania organizmu żelazem
Zaburzenia gospodarki żelazowej dzieli się następująco:
1. Genetycznie uwarunkowane przeładowanie organizmu żelazem o fenotypie hemochromatozy.
2. Niezwiązane z hemochromatozą, ale również genetycznie uwarunkowane zaburzenia gospodarki żelazowej.
3. Nabyte zespoły przeładowania żelazem.
Obecnie wyróżniamy cztery formy hemochromatozy wrodzonej, będące skutkami mutacji genetycznych genów:
- HFE
- receptora transferyny typu 2 (TfR2)
- hepcydyny (HAMP)
- hemojuweliny (HJV).
Formy wrodzone nadmiaru żelaza w organizmie niezwiązane z hemochromatozą wynikają z mutacji genów ferroportyny (FNP), ceruloplazminy i transportera metali typu 1. Trzeci typ zaburzeń towarzyszy zwykle chorobie alkoholowej wątroby, niealkoholowej stłuszczeniowej chorobie wątroby czy zakażeniom wirusami hepatotropowymi. 3
Hemochromatoza wrodzona
Termin hemochromatoza wrodzona (pierwotna lub idiopatyczna) oznacza wrodzone zaburzenie metabolizmu żelaza, które charakteryzuje się nadmiernym gromadzeniem tego pierwiastka w komórkach i tkankach z następowym ich uszkodzeniem. Choroba objawia się rozwojem zwłóknienia wątroby (z progresją do marskości narządu), endokrynopatiami i kardiomiopatią. Przeładowanie komórek żelazem wynika z wrodzonego defektu, który polega na braku hamowania napływu żelaza do krążenia, kiedy nie jest ono niezbędne. 4 Hemochromatozę jako jednostkę chorobową opisano już w XIX w., ale gen odpowiedzialny za występowanie jej najczęstszej postaci (HFE) poznano dopiero w 1996 r. Fenotyp hemochromatozy obejmuje normalną erytropoezę, podwyższoną saturację transferyny, która odzwierciedla zwiększoną osoczową pulę żelaza, oraz wysokie stężenie ferrytyny, co wskazuje na wzmożone zasoby tkankowe żelaza. 5 Inne wspólne cechy hemochromatozy to także typ dziedziczenia autosomalny recesywny, przeładowanie żelazem początkowo puli osoczowej, a następnie tkankowej z niszczeniem zajętych organów poprzez kumulację żelaza w ich tkance parenchymalnej oraz dobra odpowiedź na leczenie krwioupustami. 4 Opierając się na tej definicji, wyróżniamy:
- hemochromatozę związaną z mutacją genu HFE, którą dziedziczy się autosomalnie recesywnie i która odpowiada za >80% wszystkich rozpoznawanych przypadków tej choroby
- hemochromatozę młodzieńczą, wynikającą z mutacji genu hemojuweliny (HJV), dziedziczoną autosomalnie recesywnie
- postać związaną z mutacją genu hepcydyny (HAMP), dziedziczoną autosomalnie recesywnie
- formę hemochromatozy o przebiegu podobnym do postaci klasycznej, związaną z mutacjami genu receptora transferyny typu 2 (TfR2). 6, 7
Ponieważ najczęściej rozpoznaje się hemochromatozę związaną z mutacją genu HFE, tę formę nazywa się HFE-hemochromatozą (HFE-HC), a pozostałe – nie-HFE-hemochromatozą. Dane te podsumowywano w tabeli 1.

Tabela 1. Hemochromatoza wrodzona
Epidemiologia
Hemochromatoza wrodzona to najczęstsza choroba genetyczna rasy kaukaskiej, występująca u 1/200-250 osób. 8
Polimorfizm C282Y genu HFE (substytucja tyrozyny na cysteinę) jest nazywany dużym, gdyż większość pacjentów z hemochromatozą wrodzoną to nosiciele obu zmutowanych alleli (homozygoty C282Y genu HFE). Częstość występowania zmutowanego allelu C282Y wynosi ok. 6,2%, natomiast częstość występowania homozygotyzmu C282Y to 0,41%. Obserwowane są tu duże różnice pomiędzy różnymi regionami Europy – od 12,5% w Irlandii do blisko 0% w Europie Południowej. Nosicielami mutacji są więc częściej osoby pochodzenia celtyckiego. W Polsce 0,13% badanych to homozygoty, a 7,8% to heterozygoty C282Y genu HFE. 9 Częstość występowania zmutowanego allelu H63D (zamiana histydyny na asparaginian) wynosi ok. 14% i nie zależy w takim stopniu od regionu geograficznego, podczas gdy występowanie mutacji S65C (zamiana seryny na cysteinę) wynosi ok. 0,5% i najczęściej występuje w Wielkiej Brytanii i Francji. Spośród chorych z hemochromatozą wrodzoną aż 80,6% stanowią homozygoty C282Y genu HFE, a 5,3% to heterozygoty złożone, czyli nosiciele mutacji C282Y i H63D. U tych ostatnich często współwystępują otyłość i niealkoholowa stłuszczeniowa choroba wątroby, a ponadto stwierdza się u nich nadmierne spożywanie alkoholu. Polimorfizm S65C genu HFE może się wiązać z nadmiarem żelaza w organizmie, jeśli jest dziedziczony razem z mutacją C282Y. Dwa ostatnie polimorfizmy – H63D i S65C – traktuje się bardziej jako czynniki ryzyka przeładowania organizmu żelazem.
Objawy kliniczne
Większość chorych z klasycznym typem hemochromatozy wrodzonej szuka pomocy lekarskiej w 4 i 5 dekadzie życia, ale wczesne objawy schorzenia nie są specyficzne i obejmują: uczucie przewlekłego zmęczenia, osłabienie i bóle stawowe.
U kobiet choroba ujawnia się ok. 10 lat później, co tłumaczy się ochronnym wpływem menstruacji, ciąży i laktacji. Zaawansowana postać choroby może obejmować wiele narządów miąższowych. W przypadku wątroby możemy mieć do czynienia z dokonaną marskością, a nawet niewydolnością narządu i istotnie zwiększonym ryzykiem rozwoju pierwotnego raka wątrobowokomórkowego (HCC – hepatocellular carcinoma). To właśnie uszkodzenie wątroby wpływa na długość przeżycia chorych z najczęstszą postacią hemochromatozy wrodzonej (HFE-HC).
Problemy kliniczne obejmują także:
- kardiomiopatię objawiającą się opornymi na leczenie zaburzeniami rytmu serca oraz zastoinową niewydolnością krążenia
- endokrynopatię z cukrzycą, hipogonadyzmem hipogonadotropinowym, impotencją i niedoczynnością tarczycy
- ciemne przebarwienia na skórze (twarz, szyja, przedramiona, grzbiet rąk, okolice narządów płciowych, doły pachowe, okolice brodawek sutkowych), stąd historyczna nazwa „cukrzyca brązowa”
- artropatię pod postacią zniekształcającego zapalenia stawów o różnej lokalizacji.
Hemochromatoza związana z mutacjami genów HAMP i HJV ujawnia się znacznie wcześniej, w 2 i 3 dekadzie życia, zajęciem układu dokrewnego i serca. Chorzy umierają głównie z powodu niewydolności serca.
Typ hemochromatozy związany z mutacją genu TfR2 przebiega podobnie jak forma klasyczna schorzenia, ale ujawnia się wcześnie, chorzy zwykle w momencie jej rozpoznania mają już dokonaną marskość wątroby, cukrzycę czy kardiomiopatię. 10
Diagnostyka
Hemochromatozę wrodzoną należy podejrzewać u pacjenta z chorobą wątroby o niewyjaśnionej etiologii (np. hiperaminotransferazemią lub dokonaną marskością wątroby) oraz podwyższonym wysyceniem transferyny (>45% u kobiet, >50% u mężczyzn) i wysokim stężeniem ferrytyny (norma: >200 μg/l u kobiet i >300 μg/l u mężczyzn). Oba te parametry należy badać jednocześnie, 11 gdyż pozwalają na ocenę puli osoczowej i tkankowej żelaza, a ferrytyna dodatkowo jest białkiem ostrej fazy. Prawidłowe stężenie ferrytyny pozwala na wykluczenie hemochromatozy wrodzonej, ale podwyższenie jej stężenia w osoczu wymaga wykluczenia procesu zapalnego, zaburzeń metabolicznych, jak cukrzyca typu 2 czy choroba nowotworowa (tab. 2).

Tabela 2. Podwyższone stężenie ferrytyny - diagnostyka różnicowa

Tabela 3. Wyniki badań laboratoryjnych u pacjentów z hemochromatozą wrodzoną (wg 11 pozycji piśmiennictwa)
Stężenie żelaza w surowicy i wysycenie transferyny nie odzwierciedlają ustrojowych zasobów żelaza i nie powinny być stosowane jako markery przeładowania tkanek żelazem. 13
Mimo że hemochromatoza wrodzona jest najczęstszą chorobą metaboliczną osób rasy kaukaskiej dziedziczoną autosomalnie recesywnie (występuje 10-krotnie częściej niż mukowiscydoza), to jest rzadko rozpoznawana. Wynika to z niskiej penetracji genu, co oznacza, że u ok. 38-50% homozygot C282Y wystąpi nadmiar żelaza w organizmie, a jedynie u ok. 10-33% nosicieli obu zmutowanych alleli C282Y genu HFE choroba się rozwinie. 10 Według innych autorów ekspresja fenotypowa hemochromatozy może wystąpić u 70% homozygot C282Y, a u mniej niż 10% nosicieli obu zmutowanych alleli C282Y wystąpią objawy poważnego przeładowania organizmu żelazem z uszkodzeniem organów wewnętrznych i klinicznymi cechami hemochromatozy. 14, 15 Penetracja genu jest większa u mężczyzn niż u kobiet (28% vs 1%). 14 Powyższe dane wskazują jasno, że nie ma uzasadnienia dla wykonywania powszechnych testów genetycznych, podobnie jak poszukiwania mutacji genu HFE wśród pacjentów z dolegliwościami stawowymi czy chorych na cukrzycę typu 2. Uzasadnieniem dla testów genetycznych jest natomiast występowanie porfirii skórnej późnej, chondrokalcynozy, raka wątrobowokomórkowego czy cukrzycy typu 1. Krewni I° osób chorych na hemochromatozę wrodzoną oraz ich bliźnięta również powinni być poddani badaniu przesiewowemu z uwagi na 25% ryzyko występowania tej choroby. 10, 11, 13
Spośród badań obrazowych największe znaczenie w wykrywaniu hemochromatozy wrodzonej ma badanie metodą rezonansu magnetycznego wątroby (NMR – nuclear magnetic resonance, MRI – magnetic resonance imaging). Zaobserwowano istotną odwrotną korelację między sygnałem rezonansowym a zawartością żelaza w wątrobie, która pozwala na stwierdzenie zawartości 50-350 μmol żelaza w każdym gramie tkanki wątrobowej z czułością 84-91% i swoistością 80-100%. NMR pozwala ponadto zobrazować nierównomierne gromadzenie się zasobów żelaza w wątrobie, a także różnicować nadmierne gromadzenie żelaza w tkance parenchymalnej i mezenchymalnej czy wykrywać małe zmiany nowotworowe niezawierające żelaza. 13
Do rozpoznania hemochromatozy wrodzonej (HFE-HC) wystarcza stwierdzenie mutacji C282Y genu HFE i podwyższonych parametrów gospodarki żelazowej, niezależnie od występowania lub nie objawów klinicznych choroby.
NMR umożliwia oszacowanie zasobów żelaza w tkance parenchymalnej, a badanie metodą TK obrazuje cechy zaawansowanej marskości wątroby. Niemniej we wcześniejszych stadiach choroby biopsja wątroby odgrywa ważną rolę w ocenie stopnia zwłóknienia narządu. Należy ją wykonywać u chorych >40 r.ż., z hepatomegalią, podwyższoną aktywnością aminotransferazy asparaginianowej (AspAT – aspartate aminotransferase) lub alaninowej (AlAT – alanine aminotransferase) oraz stężeniem ferrytyny >1000 μg/l. 11, 13 Stężenie ferrytyny <1000 μg/l oraz prawidłowa aktywność AspAT, przy braku hepatomegalii, pozwala wykluczyć zaawansowane włóknienie wątroby z 95% prawdopodobieństwem. 13 Osoczowe stężenie ferrytyny można uznać za pojedynczy marker zaawansowanego włóknienia wątroby u pacjentów nosicieli obu zmutowanych alleli C282Y genu HFE. 11 Wskazaniem do biopsji może być również podwyższenie parametrów gospodarki żelazowej lub kliniczne objawy przeładowania organizmu żelazem u pacjentów, u których nie stwierdzono mutacji C282Y czy H63D. 11 Kwas hialuronowy oraz elastografia również mogą być przydatne w ocenie zaawansowanego włóknienia wątroby, ale nie są jeszcze szeroko stosowane w diagnostyce. 16, 17
Na rycinie 1 przedstawiono algorytm diagnostyczno-leczniczy w hemochromatozie wrodzonej według zaleceń AASLD.
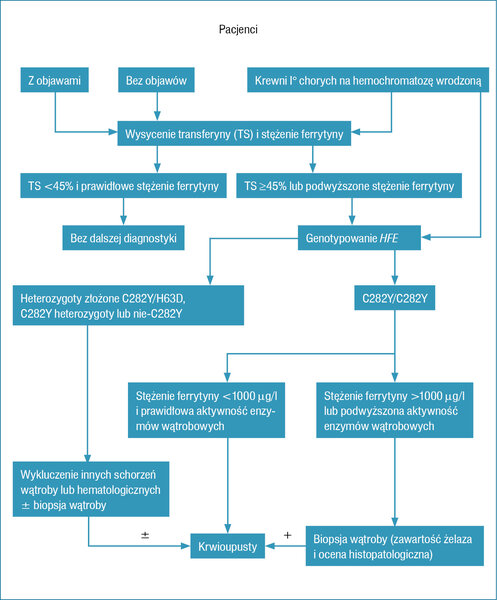
Rycina 1. Algorytm postępowania diagnostyczno-leczniczego w hemochromatozie wątrobowej (wg 11 pozycji piśmiennictwa)
Leczenie
Nie ma, niestety, danych jednoznacznie wskazujących na właściwy moment włączenia terapii hemochromatozy wrodzonej, gdyż nie określono stężenia żelaza, od którego rozpoczyna się uszkadzanie komórek i tkanek ani nie wykazano związku między zawartością żelaza w wątrobie, stężeniem ferrytyny a stopniem uszkodzenia wątroby. Najpowszechniejszym sposobem leczenia są krwioupusty. Ewakuacja 500 ml krwi pełnej pozwala na usunięcie ok. 250 mg żelaza. Związki chelatujące żelazo (deferoksamina, deferazyroks) ze względu na niższą skuteczność (jednorazowo usuwają ok. 100 mg żelaza z organizmu) stosuje się u chorych, którzy nie tolerują krwioupustów bądź nie mogą być im poddawani. Wskazaniami do ich wdrożenia są zespoły dyserytropoetyczne i przewlekła niedokrwistość hemolityczna. 11 Najrzadziej stosowaną metodą leczenia jest erytrocytofereza. 13
Rozpoczęcie leczenia krwioupustami, zanim dojdzie do rozwoju marskości wątroby czy cukrzycy, znacząco obniża chorobowość i śmiertelność w przebiegu hemochromatozy wrodzonej. Zaleca się więc włączenie terapii u chorych bez objawów, którzy są homozygotami C282Y genu HFE i stwierdza się u nich podwyższone parametry gospodarki żelazowej (ferrytyna >1000 μg/l), a także u pacjentów z objawami choroby czy cechami uszkodzenia narządów miąższowych. Postępowanie takie jest zasadne m.in. z uwagi na to, że HCC zdecydowanie rzadziej się rozwija u chorych bez dokonanej marskości wątroby. Ponieważ HCC jest przyczyną ok. 30% zgonów u chorych na hemochromatozę wrodzoną, a powikłania marskości wątroby 20% zgonów, u pacjentów z marskością wątroby w przebiegu hemochromatozy wrodzonej należy regularnie, co 6 miesięcy, wykonywać badanie USG jamy brzusznej. 11 Kwestię dyskusyjną stanowi natomiast terapia krwioupustami u chorych będących nosicielami obu zmutowanych alleli C282Y genu HFE, z prawidłową aktywnością enzymów wątrobowych, bez objawów klinicznych oraz z miernie podwyższonym stężeniem ferrytyny (<1000 μg/l). Wydaje się, że mimo braku przekonujących danych z badań i publikacji należy jednak rozważać u nich taką metodę leczenia. 11
Krwioupusty powinny być prowadzone początkowo raz w tygodniu (niemniej w zależności od tolerancji chorego możliwe jest wydłużenie odstępu czasowego do jednego razu na 2 tygodnie), do czasu osiągnięcia saturacji transferyny <45-50% oraz, przede wszystkim, stężenia ferrytyny 50-100 μg/l lub wystąpienia anemizacji (obniżenia hematokrytu/hemoglobiny <80% poziomu wyjściowego). 11 Następnie krwioupusty prowadzi się w ramach leczenia podtrzymującego, zwykle raz na 3-6 miesięcy, i kontroluje stężenie ferrytyny co ok. 3 miesiące. Nie ma jednak dostatecznej liczby badań, które pozwoliłyby na stworzenie protokołu wenesekcji opartego na faktach, a powyższe dane wynikają z doświadczenia zajmujących się taką terapią ośrodków i opierają się na dostępnym piśmiennictwie. 10, 11, 13
Skuteczność terapii krwioupustami zależy od stopnia uszkodzenia narządów w momencie rozpoczęcia leczenia. Zwykle poprawia się stopień przewlekłego zmęczenia, zmniejsza się natężenie bólów brzucha i nadmierna pigmentacja skóry, spada aktywność aminotransferaz, a także poprawia się kontrola glikemii. Obserwowano również regresję włóknienia wątroby, a nawet zmniejszanie się żylaków przełyku u chorych z dokonaną marskością wątroby. 18, 19 Krwioupusty najczęściej jednak nie wpływają na dolegliwości stawowe czy ryzyko rozwoju HCC u chorych z dokonaną marskością wątroby w przebiegu hemochromatozy wrodzonej, a wpływ na układ dokrewny czy serce zależy od wyjściowego stopnia ich uszkodzenia. 13 U chorych, u których wcześnie rozpoczęto leczenie, czas przeżycia będzie zbliżony do tego w populacji ogólnej.
Niewydolność wątroby z wodobrzuszem, encefalopatią, krwawieniami z żylaków przełyku czy rozwój HCC są wskazaniami do rozważenia przeszczepienia wątroby. Publikowane wyniki były jednak nieco gorsze niż w przypadku transplantacji z innych wskazań: rok przeżywało 64% pacjentów (podczas gdy średnie roczne przeżycie u chorych po transplantacjach wykonanych w Europie po 1999 r. wynosiło ok. 84%), a pięć lat ok. 34% (w innych wskazaniach średnio 75%), co wiązało się najprawdopodobniej z wyjściowym uszkodzeniem innych narządów i większym odsetkiem osób z niewydolnością serca, zakażeniami i chorobami nowotworowymi. 20 Obecnie jednak wyniki przeszczepienia wątroby poprawiają się i stają się zbliżone do uzyskiwanych w innych wskazaniach, co zapewne wynika ze skuteczniejszego leczenia chorych przed zabiegiem, szczególnie krwioupustami. 21
W zaleceniach dietetycznych dla chorych poddawanych regularnym krwioupustom zawarto jedynie ograniczenia w stosowaniu suplementów wielowitaminowych zawierających żelazo, a także witaminy C w dawce przekraczającej 500 mg/24 h. Picie herbaty prawdopodobnie obniża stężenie żelaza zapasowego u chorych z hemochromatozą wrodzoną, 22 chociaż nie potwierdzono tego w innych badaniach. 23 Pacjenci powinni także stosować ogólnie przyjęte zalecenia dotyczące rozsądnej diety i trybu życia. Alkohol nasila uszkodzenie wątroby w mechanizmie stresu oksydacyjnego, intensyfikuje wchłanianie żelaza w jelicie cienkim oraz hamuje wątrobową ekspresję hepcydyny, dlatego chorym z hemochromatozą wrodzoną zaleca się ograniczenie jego spożywania. 13
Podsumowanie
Hemochromatoza wrodzona (HC) jest dziedziczonym autosomalnie recesywnie schorzeniem, w przebiegu którego przy prawidłowej erytropoezie dochodzi do kumulacji żelaza w tkance parenchymalnej wątroby, serca i gruczołów wydzielania wewnętrznego. Proces ten może prowadzić do zaawansowanego uszkodzenia tych narządów. Główną przyczyną choroby, u ok. 80% osób, jest mutacja C282Y genu HFE, który jest regulatorem hepcydyny – najważniejszego hormonu regulującego gospodarkę żelazową u człowieka. Mutacje innych genów regulatorowych są rzadsze (nie-HFE-hemochromatoza). Mimo częstego występowania mutacji C282Y u ludzi rasy białej objawy hemochromatozy rozwijają się rzadko, co wynika z niskiej penetracji genu. Do rozpoznania choroby wymagane jest stwierdzenie podwyższonej saturacji transferyny i wysokiego stężenia ferrytyny oraz homozygotycznego genotypu C282Y genu HFE. Badania obrazowe, np. NMR, oraz biopsja wątroby pozwalają na ocenę depozytów żelaza i zaawansowania włóknienia wątroby. Leczenie polega na stosowaniu powtarzanych krwioupustów, prowadzonych do czasu osiągnięcia prawidłowych parametrów gospodarki żelazowej. Następnie wykonuje się je w ramach terapii podtrzymującej przez całe życie chorego. Wskazane jest także istotne ograniczenie spożycia alkoholu.
- 1. Nemeth E, Tuttle MS, Powelson J, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004;306(5704):2090-3.
- 2. Nemeth E, Ganz T. The role of hepcidin in iron metabolism. Acta Haematol 2009;122(2-3):78-86.
- 3. Deugnier Y, Brissot P, Loreal O. Iron and the liver: update 2008. J Hepatol 2008;48(Suppl 1):S113-23.
- 4. Pietrangelo A. Molecular insights into the pathogenesis of hereditary haemochromatosis. Gut 2006;55(4):564-8.
- 5. Pietrangelo A. Hemochromatosis: an endocrine liver disease. Hepatology 2007;46(4):1291-301.
- 6. Camaschella C, Roetto A, Cali A, et al. The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22. Nat Genet 2000;25(1):14-5.
- 7. Papanikolaou G, Samuels ME, Ludwig EH, et al. Mutations in HFE2 cause iron overload in chromosome 1q-linked juvenile hemochromatosis. Nat Genet 2004;36(1):77-82.
- 8. Phatak PD, Bonkovsky HL, Kowdley KV. Hereditary hemochromatosis: time for targeted screening. Ann Intern Med 2008;149(4):270-2.
- 9. Raszeja-Wyszomirska J, Kurzawski G, Suchy J, et al. Frequency of mutations related to hereditary haemochromatosis in northwestern Poland. J Appl Genet 2008;49(1):105-7.
- 10. Pietrangelo A. Hereditary hemochromatosis: pathogenesis, diagnosis, and treatment. Gastroenterology 2010;139(2):393-408.
- 11. Bacon BR, Adams PC, Kowdley KV, et al. Diagnosis and management of hemochromatosis: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology 2011;54(1):328-43.
- 12. Asberg A, Hveem K, Kannelonning K, et al. Penetrance of the C28Y/C282Y genotype of the HFE gene. Scand J Gastroenterol 2007;42(9):1073-7.
- 13. EASL clinical practice guidelines for HFE hemochromatosis. J Hepatol 2010;53(1):3-22.
- 14. Allen KJ, Gurrin LC, Constantine CC, et al. Iron-overload-related disease in HFE hereditary hemochromatosis. N Engl J Med 2008;358(3):221-30.
- 15. Beutler E, Felitti VJ, Koziol JA, et al. Penetrance of 845G--> A (C282Y) HFE hereditary haemochromatosis mutation in the USA. Lancet 2002;359(9302):211-8.
- 16. Adhoute X, Foucher J, Laharie D, et al. Diagnosis of liver fibrosis using FibroScan and other noninvasive methods in patients with hemochromatosis: a prospective study. Gastroenterol Clin Biol 2008;32(2):180-7.
- 17. Crawford DH, Murphy TL, Ramm LE, et al. Serum hyaluronic acid with serum ferritin accurately predicts cirrhosis and reduces the need for liver biopsy in C282Y hemochromatosis. Hepatology 2009;49(2):418-25.
- 18. Falize L, Guillygomarc'h A, Perrin M, et al. Reversibility of hepatic fibrosis in treated genetic hemochromatosis: a study of 36 cases. Hepatology 2006;44(2):472-7.
- 19. Fracanzani AL, Fargion S, Romano R, et al. Portal hypertension and iron depletion in patients with genetic hemochromatosis. Hepatology 1995;22(4 Pt 1):1127-31.
- 20. Kowdley KV, Brandhagen DJ, Gish RG, et al. Survival after liver transplantation in patients with hepatic iron overload: the national hemochromatosis transplant registry. Gastroenterology 2005;129(2):494-503.
- 21. Yu L, Ioannou GN. Survival of liver transplant recipients with hemochromatosis in the United States. Gastroenterology 2007;133(2):489-95.
- 22. Kaltwasser JP, Werner E, Schalk K, et al. Clinical trial on the effect of regular tea drinking on iron accumulation in genetic haemochromatosis. Gut 1998;43(5):699-704.
- 23. Milward EA, Baines SK, Knuiman MW, et al. Noncitrus fruits as novel dietary environmental modifiers of iron stores in people with or without HFE gene mutations. Mayo Clin Proc 2008;83(5):543-9.
Następny artykuł:


 Dodaj do ulubionych
Dodaj do ulubionych
