


Spis treści
Gromadzenie się metali w ośrodkowym układzie nerwowym prowadzi do ciężkich i często nieodwracalnych uszkodzeń. Toksyczny wpływ metali na ten układ może wynikać z egzogennego zatrucia związkami zawierającymi metale (np. efedron), jak również mieć podłoże genetyczne. Najlepiej poznanymi genetycznymi chorobami neurologicznymi są zespoły spowodowane gromadzeniem się żelaza, manganu i miedzi.
Wprowadzenie
W artykule omówiono trzy grupy chorób. Pierwszą jest neurodegeneracja z gromadzeniem żelaza
(NBIA – neurodegeneration with brain iron accumulation), obejmująca jednostki chorobowe z różnorodnymi zaburzeniami neurologicznymi. Do grupy drugiej należy zespół chorobowy związany z gromadzeniem manganu, charakteryzujący się uszkodzeniem wątroby, policytemią oraz uogólnioną dystonią. Wreszcie choroba Wilsona, związana z zaburzeniem metabolizmu miedzi, cechuje się uszkodzeniem wątroby oraz objawami neuropsychiatrycznymi.
Neurodegeneracja z gromadzeniem żelaza
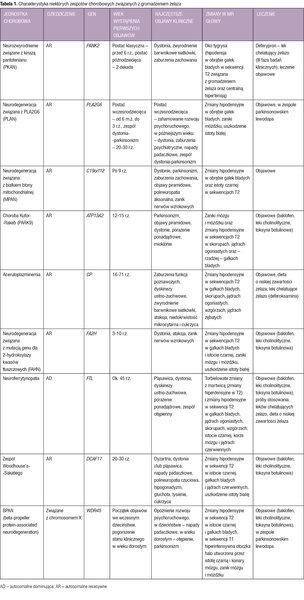
Tabela 1. Charakterystyka niektórych zespołów chorobowych związanych z gromadzeniem żelaza
Poniżej omówiono najczęściej występujące zespoły NBIA. Pozostałe, bardzo rzadkie jednostki chorobowe związane z gromadzeniem żelaza, m.in. chorobę Kufor-Rakeb, aceruloplazminemię, neurodegenerację związaną z mutacją genu dla 2-hydroksylazy kwasów tłuszczowych (FAHN – fatty acid hydroxylase-associated neurodegeneration), neuroferrytynopatię i BPAN (beta-propeller protein-associated neurodegeneration), przedstawiono w tabeli 1. 1, 2, 3, 4, 5, 6, 7, 8
Neurozwyrodnienie związane z kinazą pantotenianu
Neurozwyrodnienie związane z kinazą pantotenianu (PKAN – pantothenate kinase-associated neurodegeneration) to najczęściej występujący zespół (1-3/1 mln przypadków). 1, 2, 3, 4 Jego przyczyną jest mutacja w genie kodującym kinazę pantotenianu 2, która bierze udział w biosyntezie koenzymu A, niezbędnego do metabolizmu kwasów tłuszczowych. Skutkuje to zaburzeniami procesów β-oksydacji kwasów tłuszczowych i odkładaniem się L-cysteiny chelatującej żelazo. 1, 2, 3, 4
W postaci klasycznej PKAN pierwsze objawy występują przed 6 r.ż. Dominuje sztywność mięśniowa, stopniowo rozwija się uogólniona dystonia. Ponadto stwierdza się zwyrodnienie barwnikowe siatkówki oraz obecność akantocytów w rozmazie krwi. W postaci późnodziecięcej objawy pojawiają się w 2 dekadzie życia i są mniej nasilone niż w postaci klasycznej. Często występują zaburzenia zachowania. Choroba o początku objawów w wieku dorosłym cechuje się przedłużonym przebiegiem. 1, 2, 3, 4
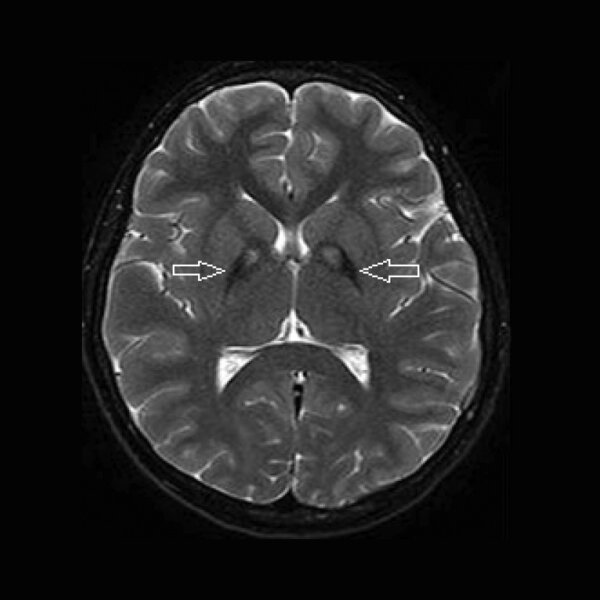
Rycina 1. MR głowy, obrazy T2-zależne. Objaw oka tygrysa u pacjenta z PKAN (zmiany hipodensyjne w obrębie gałek bladych związane z gromadzeniem się żelaza oraz centralna hiperdensja)
Charakterystyczne dla PKAN są zmiany w MR głowy. Stwierdzenie objawu oka tygrysa (hipodensja w obrębie gałek bladych w sekwencji T2 związana z gromadzeniem żelaza oraz centralną hipertensją) może być bardzo pomocne w diagnostyce choroby (ryc. 1). Rozmaz krwi obwodowej wykazuje obecność akantocytów. Istotną rolę w diagnostyce odgrywa także badanie genetyczne, które może potwierdzić rozpoznanie. 1, 2, 3, 4
W leczeniu choroby wykorzystuje się deferypron, lek chelatujący żelazo. Wyniki terapii nie zostały w pełni potwierdzone badaniami klinicznymi – dostępne są tylko obserwacje przypadków. 9, 10 W przypadku PKAN stosuje się obecnie leczenie objawowe. Chorym podaje się lewodopę, toksynę botulinową, baklofen i benzodiazepiny. Podejmowane są również próby leczenia głęboką stymulacją mózgu (DBS – deep brain stimulation), z różnymi efektami. 1, 2, 3, 4
Neurodegeneracja związana z PLA2G6
Neurodegeneracja związana z PLA2G6 (PLAN – PLA2G6-associated neurodegeneration) to drugi co do częstości występowania zespół NBIA. Jego przyczyną jest mutacja w genie kodującym białko PLA2G6, znajdującym się na chromosomie 22. PLA2G6 to fosfolipaza zależna od wapnia, zaangażowana w metabolizm wolnych kwasów tłuszczowych. 1, 2, 3, 4 W PLAN dochodzi do zaburzenia przepuszczalności błon komórkowych.
Wyróżnia się trzy główne postaci choroby: dystrofię neuroaksonalną Seitelbergera (postać wczesnodziecięca), atypową dystrofię neuroaksonalną (ANAD – atypical neuroaxonal dystrophy) i zespół dystonia-parkinsonizm (PARK14). 1, 2, 3, 4 Postać wczesnodziecięca przebiega z zahamowaniem rozwoju psychoruchowego oraz napadami padaczkowymi, a śmierć następuje w ciągu kilku lat. W ANAD objawy pojawiają się nieco później. Najczęściej obserwuje się dystonię, zaburzenia psychiatryczne oraz napady padaczkowe. Zespół dystonia-parkinsonizm rozpoczyna się w wieku 20-30 lat i charakteryzuje dobrą odpowiedzią na lewodopę. MR mózgu wykazuje odkładanie się żelaza w gałkach bladych, zaniki móżdżku i uszkodzenie istoty białej. Badanie genetyczne potwierdza rozpoznanie. Leczenie zespołu jest wyłącznie objawowe. 1, 4, 6
Neurodegeneracja związana z białkiem błony mitochondrialnej
Za neurodegenerację związaną z białkiem błony mitochondrialnej (MPAN – mitochondrial membrane protein-associated neurodegeneration) odpowiada gen C19orf12, który znajduje się na chromosomie 19. Objawy pojawiają się stopniowo po 9 r.ż. Często obserwuje się dystonie, parkinsonizm, zaburzenia zachowania i objawy piramidowe. Ponadto występują polineuropatia aksonalna oraz zanik nerwów wzrokowych. MR mózgu wykazuje zmiany hipodensyjne w obrębie gałek bladych oraz istoty czarnej w sekwencjach T2, natomiast nie opisuje się klasycznego oka tygrysa w tym badaniu. Do potwierdzenia rozpoznania służy badanie genetyczne. Leczenie jest tylko objawowe. 1, 2, 3, 4, 8
Choroby związane z gromadzeniem manganu
Bardzo rzadka choroba genetyczna związana z gromadzeniem manganu spowodowana jest mutacją w genie SLC30A10, który koduje transporter manganu. Skutkuje to odkładaniem się manganu głównie w jądrach podstawy i w móżdżku. 11, 12
Pierwsze objawy pod postacią dystonii uogólnionej zwykle pojawiają się w 1 dekadzie życia. Występują również zaburzenia mowy o typie dyzartrii, zespół parkinsonowski, neuropatia ruchowa oraz zaburzenia psychiatryczne. Charakterystycznym objawem jest marskość wątroby. W preparatach z biopsji wątroby stwierdza się podwyższone stężenie manganu, a w badaniach laboratoryjnych – policytemię oraz obniżone stężenie ferrytyny i żelaza w surowicy. MR głowy w sekwencji T1 wykazuje hiperintensywne zmiany zlokalizowane głównie w gałkach bladych, a także w prążkowiu, móżdżku i istocie białej. 11, 12
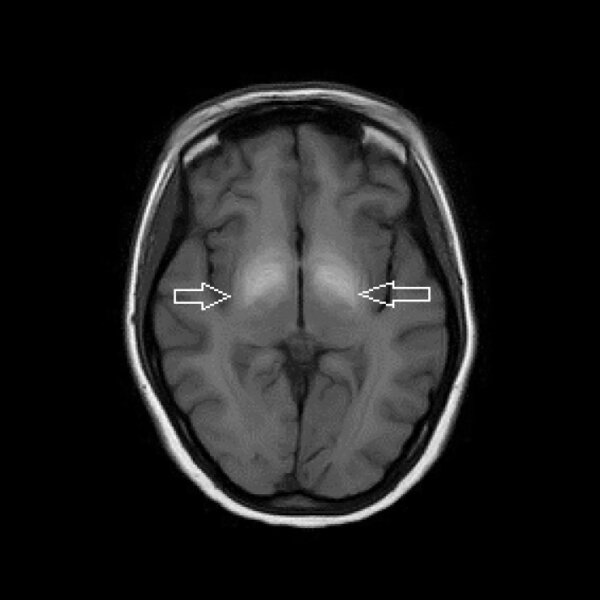
Rycina 2. MR głowy. Hiperintensywne zmiany w obrazach T1-zależnych w gałkach bladych związane z gromadzeniem się manganu u pacjentki nadużywającej efedronu
W chorobie związanej z gromadzeniem manganu stosuje się leczenie chelatujące z wykorzystaniem soli wapniowo-disodowej kwasu wersenowego, prowadzące do wzmożonego wydalania manganu. W większości przypadków następuje poprawa kliniczna. Dodatkowo prowadzi się suplementację preparatów żelaza, ponieważ żelazo stabilizuje stężenie manganu w surowicy. 11, 12
Przyczyną odkładania się manganu w ośrodkowym układzie nerwowym (OUN) może być również nadużywanie efedronu (ryc. 2). Efedron (metylokatynon) jest inhibitorem wychwytu zwrotnego noradrenaliny oraz dopaminy i działa jako psychostymulant. Dodatek stosowany do sporządzania roztworu do iniekcji dożylnych zawiera nadmanganian potasu. W tak uzyskanym roztworze znajduje się duża ilość manganu, który odkłada się w OUN, głównie w jądrach podkorowych. W diagnostyce różnicowej należy uwzględnić nabyte zwyrodnienie wątrobowo-soczewkowe w schyłkowej niewydolności wątroby. 5
Choroba Wilsona
Choroba Wilsona to rzadka choroba dziedziczona autosomalnie recesywnie, w której dochodzi do zaburzenia metabolizmu miedzi i jej odkładania się w różnych narządach, głównie w wątrobie i mózgu. Najczęściej u chorych obserwuje się objawy uszkodzenia wątroby, objawy neurologiczne i psychiatryczne. 13, 14, 15 Przyczyną zaburzeń metabolizmu miedzi są mutacje w genie ATP7B zlokalizowanym na chromosomie 13. 14 Gen ten koduje białko ATP-azę, która pośredniczy we wbudowywaniu miedzi w apoceruloplazminę. Na skutek nieprawidłowego funkcjonowania ATP-azy miedź gromadzi się w hepatocytach, uszkadza je, następnie jest uwalniana do krwiobiegu i powoduje uszkodzenie kolejnych narządów: mózgu, rogówki, nerek oraz serca. 14
Objawy kliniczne
Pierwsze symptomy choroby obserwuje się najczęściej między 10 a 40 r.ż. Obraz kliniczny jest zróżnicowany: występują objawy uszkodzenia wątroby, neurologiczne oraz psychiatryczne, 13, 14, 15 przy czym objawy neurologiczne zwykle rozwijają się później niż objawy uszkodzenia wątroby. Te ostatnie mogą się ujawniać jako wzrost aktywności enzymów wątrobowych, bezobjawowe powiększenie wątroby i śledziony. Występują także ostre i przewlekłe zapalenie wątroby oraz marskość. Wśród objawów neurologicznych dominują zaburzenia mowy, drżenie (pozycyjne, zamiarowe, spoczynkowe), dystonia i zaburzenia chodu.
Choroba Wilsona daje dwa charakterystyczne objawy: drżenie przypominające bicie skrzydłami, które dotyczy proksymalnych części kończyn górnych, 16 oraz pierścień Kaysera-Fleischera, będący efektem odkładania się miedzi w przednich warstwach rogówki oka na błonie Descemeta. 13 Poza opisanymi najczęściej występującymi objawami można zaobserwować również zaburzenia hematologiczne, endokrynologiczne, kostno-stawowe, nefrologiczne i kardiologiczne. 13, 14
Dla celów praktycznych została opracowana skala do oceny stanu neurologicznego pacjentów z chorobą Wilsona, pozwalająca ocenić nasilenie objawów neurologicznych i stan funkcjonalny. 17
Diagnostyka
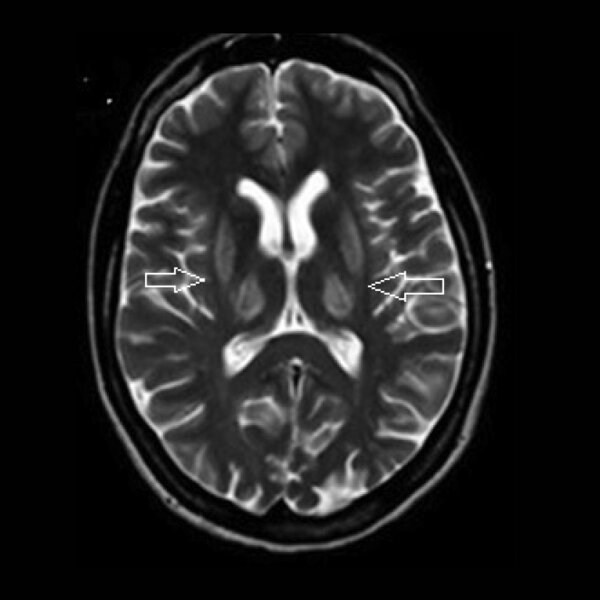
Rycina 3. MR głowy. Hiperintensywne zmiany w obrazach T2-zależnych w skorupach i we wzgórzach u pacjentki z chorobą Wilsona
Rozpoznanie opiera się na kombinacji objawów klinicznych, wyników badań laboratoryjnych i neuroobrazowych oraz identyfikacji mutacji w genie ATP7B. 13, 14, 15 W przypadku podejrzenia choroby Wilsona należy wykonać badania metabolizmu miedzi: oznaczenie stężenia ceruloplazminy (zwykle obniżone o więcej niż 50% dolnej granicy normy) i miedzi całkowitej w surowicy (na ogół obniżone) oraz badanie wydalania miedzi w dobowej zbiórce moczu (najczęściej podwyższone). U osób chorych wydalanie miedzi w dobowej zbiórce moczu wzrasta do ponad 100 g/24 h. 13 W niektórych przypadkach stosuje się również test z wbudowywaniem miedzi radioaktywnej do ceruloplazminy. 15
W diagnostyce choroby Wilsona dużą rolę odgrywa badanie genetyczne, które służy identyfikacji mutacji w genie ATP7B. 14 Istotne jest także potwierdzenie obecności charakterystycznego dla tej choroby pierścienia Kaysera-Fleischera, obserwowanego u większości pacjentów z zaburzeniami neurologicznymi. 15 Prawie u wszystkich chorych z objawami neurologicznymi MR głowy pokazuje nieprawidłowości. 18 Zmiany występują zazwyczaj w jądrach podstawy: w skorupach, jądrach ogoniastych, wzgórzach, śródmózgowiu, moście (ryc. 3). Przeważnie obserwuje się zmiany hiperintensywne, rzadziej hipointensywne w sekwencji T2. Często występuje zanik mózgu i móżdżku. 18
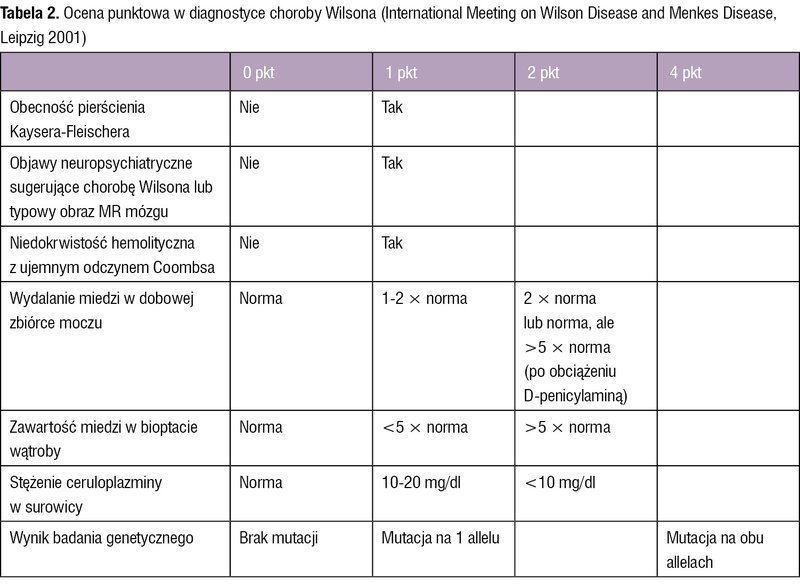
Tabela 2. Ocena punktowa w diagnostyce choroby Wilsona (International Meeting on Wilson Disease and Menkes Disease, Leipzig 2001)
Leczenie i jego monitorowanie
Bardzo ważne jest wczesne rozpoznanie choroby i włączenie leczenia przeciwmiedziowego. Jeśli nie podejmie się terapii, postępuje uszkodzenie wątroby i narastają objawy neurologiczne. Zwykle kończy się to zgonem. Celem leczenia jest usunięcie nadmiaru miedzi z organizmu i zapobieganie jej reakumulacji. Wczesne rozpoznanie oraz regularne przyjmowanie leków przeciwmiedziowych przez całe życie może poprawić stan kliniczny lub nawet doprowadzić do całkowitego wycofania objawów klinicznych. W przypadkach z zaawansowanymi zmianami w mózgu i/lub z szybko postępującą niewydolnością wątroby leczenie może być nieskuteczne.
W Polsce w terapii choroby Wilsona stosowane są dwa leki: D-penicylamina oraz cynk. D-penicylamina jest lekiem chelatującym miedź i powoduje wydalanie jej z moczem. 13, 15, 20, 21 Lek podaje się w dawce 1,0-1,5 g/24 h, zwykle w 3 dawkach podzielonych. 14 Aby zapewnić jak najlepsze wchłanianie, D-penicylaminę należy przyjmować 1 h przed posiłkiem lub 2 h po posiłku. 13 Wprowadza się ją bardzo powoli (1/2 tabletki co kilka dni), ponieważ istnieje ryzyko pogorszenia stanu neurologicznego (10-50% pacjentów). Prawidłowość leczenia ocenia się, monitorując parametry metabolizmu miedzi, szczególnie stężenie wydalania miedzi w dobowej zbiórce moczu, które w przypadku leków chelatujących powinno być zwiększone. 13
Drugim lekiem dostępnym w Polsce do leczenia choroby Wilsona jest cynk, który zapobiega wchłanianiu miedzi z przewodu pokarmowego. Obecnie sole cynku są zalecane głównie u pacjentów presymptomatycznych lub w terapii podtrzymującej, 14 jednak według badań prowadzonych przez II Klinikę Neurologiczną skuteczność soli cynku i D-penicylaminy w terapii początkowej jest porównywalna. 20, 21 Proponowane dawki cynku w leczeniu choroby Wilsona wynoszą od 150 do 250 mg/24 h, podawane w 3 dawkach. 13, 14, 15
Monitorowanie leczenia farmakologicznego opiera się na regularnej ocenie neurologicznej pacjentów oraz ocenie parametrów funkcji wątroby. 15 W przypadku leczenia D-penicylaminą wydalanie miedzi w dobowej zbiórce moczu powinno mieścić się w zakresie 200-500 µg, a w przypadku leczenia solami cynku, które ograniczają wchłanianie miedzi z przewodu pokarmowego – wynosić mniej niż 100 µg/24 h. 13, 14, 15
Przeszczepienie wątroby jest postępowaniem zarezerwowanym dla osób z chorobą Wilsona rozwijających piorunującą niewydolność wątroby oraz dla pacjentów z ciężką zdekompensowaną marskością wątroby, niereagującą na leczenie lekami chelatującymi. 22
Badania przesiewowe krewnych pacjentów
Ponieważ choroba Wilsona jest dziedziczona autosomalnie recesywnie, najwyższe ryzyko zachorowania (25%) dotyczy rodzeństwa pacjenta. Teoretycznie ryzyko u potomstwa chorych jest stosunkowo niskie: 13 wśród dzieci pacjentów II Kliniki Neurologicznej choroba Wilsona została stwierdzona u ok. 4%. 23 Często jednak ustalenie rozpoznania u krewnych bywa trudne, ponieważ u chorych bez objawów wyniki badań metabolizmu są w granicach normy lub tylko nieznacznie zaburzone. Niewielkie zmiany w metabolizmie miedzi można stwierdzić również u heterozygot (osób z mutacją w jednym allelu genu ATP7B). Pomocne jest badanie genetyczne, zwłaszcza w diagnostyce choroby u rodzeństwa pacjenta ze znaną mutacją. Zarówno wytyczne europejskie, jak i amerykańskie zalecają badanie krewnych pierwszego stopnia. 14, 15
Podsumowanie
NBIA to grupa chorób z różnorodnymi zaburzeniami neurologicznymi (ruchy mimowolne, parkinsonizm, ataksje, zaburzenia poznawcze) oraz hipodensyjnymi zmianami w obrębie jąder podstawy w MR głowy w sekwencji T2. Większość zespołów chorobowych rozpoczyna się w dzieciństwie, ale niektóre również w późniejszym wieku. Do potwierdzenia rozpoznania służy badanie genetyczne. W większości zespołów NBIA leczenie jest objawowe.
Rzadki zespół chorobowy uwarunkowany genetycznie (mutacja w genie SLC30A10), związany z gromadzeniem się manganu w jądrach podstawy, cechuje się uszkodzeniem wątroby oraz obecnością objawów neurologicznych (uogólniona dystonia). W MR głowy w sekwencji T1 obserwuje się hiperintensywne zmiany zlokalizowane w jądrach podstawy i móżdżku. Leczenie opiera się na preparatach chelatujących mangan i żelazo, które u większości pacjentów są skuteczne.
W chorobie Wilsona, polegającej na zaburzeniu metabolizmu miedzi, najczęściej występują uszkodzenie wątroby oraz objawy neurologiczne (zaburzenia mowy, drżenie, zaburzenia chodu, dystonia) i psychiatryczne. Rozpoznanie opiera się na kombinacji objawów klinicznych i wyników badań laboratoryjnych, stwierdzeniu obecności pierścienia Kaysera-Fleischera oraz identyfikacji mutacji w genie ATP7B. MR pokazuje zmiany zazwyczaj w jądrach podstawy: w skorupach, jądrach ogoniastych, wzgórzach, śródmózgowiu, moście. Chorobę Wilsona można skutecznie leczyć lekami przeciwmiedziowymi.
Wiedza o patogenezie i diagnostyce genetycznie uwarunkowanych chorób OUN przebiegających z gromadzeniem metali jest coraz większa, co najprawdopodobniej przełoży się w przyszłości na lepsze możliwości terapeutyczne tych jednostek chorobowych.
Abstract
Genetic CNS disorders associated with accumulation of metals (iron, manganese and copper) – diagnostic and therapeutic algorithms
The accumulation of metals in the central nervous system is caused by genetic or environmental factors and may result in severe and often irreversible impairment. The most common genetic syndromes associated with accumulation of metals include iron, manganese, and copper deposition. Neurodegeneration with brain iron accumulation (NBIA) is a clinically and genetically heterogeneous group of progressive disorders characterized by elevated levels of brain iron. Most of them are inherited in an autosomal recessive manner. They are characterized by a wide range of neurological signs and symptoms, including dystonia, parkinsonism, ataxia and dementia. The first symptoms in most patients occur in childhood. A characteristic magnetic resonance imaging (MRI) sign of iron deposition is the presence of hypointense signals on T2-weighted images. Genetic testing may help in differential diagnosis. In some NBIA syndromes, iron depletion therapy is successful (aceruloplasminemia), but in others effective therapy is still lacking. Hypermanganesemia with genetic etiology is caused by mutations in the SLC30A10 gene, encoding a manganese transporter. The main signs of this rare syndrome involve early-onset generalized dystonia and hepatic cirrhosis, dystonia and polycythemia. Treatment with CaNa2-EDTA has been effective in most of these patients. Wilson’s disease (WD) is a rare disorder of copper metabolism. WD is an autosomal recessive inherited disorder and is caused by mutations in ATP7B gene. The most common symptoms are hepatic, neurologic, and psychiatric. Diagnosis is based on clinical symptoms, copper metabolism tests, the presence of a Kayser-Fleischer ring in the cornea and genetic testing. WD is treatable disorder with anti-copper agents. In most cases clinical improvement is seen. However, untreated WD may lead to death.
- 1. Kruer MC, Boddaert N. Neurodegeneration with brain iron accumulation: a diagnostics algorithm. Semin Pediatr Neurol 2012;19:67-74.
- 2. Schneider SA, Bhatia KP. Syndromes of neurodegeneration with brain iron accumulation. Semin Pediatr Neurol 2012;19:57-66.
- 3. Dusek P, Schneider SA. Neurodegeneration with brain iron accumulation. Curr Opin Neurol 2012;25:499-506.
- 4. Schneider SA, Hardy J, Bhatia K. Syndromes of neurodegeneration with brain iron accumulation (NBIA) and update on clinical presentations, histological and genetic underpinnings, and treatment considerations. Mov Disord 2012;27:42-52.
- 5. Dusek P, Litwin T, Członkowska A. Wilson disease and other neurodegenerations with metal accumulations. Neurologic Clinics 2015;33:175-204.
- 6. Kalman B, Lautenschlaeger R, Kohlmyer F, et al. An international registry for neurodegeneration with brain iron accumulation. Orphanet J Rare Dis 2012;7:66.
- 7. Morgan NV, Westaway SK, Morton JE, et al. PLA2G6, encoding phospholipase A2, is mutated in neurodegenerative disorders with high brain iron. Nat Genet 2006;38:752-54.
- 8. Deschauer M, Gaul C, Behrmann C, et al. C19orf12 mutations in neurodegeneration with brain iron accumulation mimicking juvenile amyotrophic lateral sclerosis. J Neurol 2012;259:2434-9.
- 9. Zorzi G, Zibordi F, Chiapparini L, et al. Iron-related MRI images in patients with pantothenate kinase-associated neurodegeneration (PKAN) treated with deferiprone: results of a phase II pilot trial. Mov Disord 2011;26:1755-9.
- 10. Kwiatkowski A, Ryckewaert G, Jissendi Tchofo P, et al. Long-term improvement under deferiprone in a case of neurodegeneration with brain iron accumulation. Parkinsonism and Relat Disord 2012;18:110-2.
- 11. Tuschl K, Clayton PT, Gospe SM Jr, et al. Syndrome of hepatic cirrhosis, dystonia, polycythemia, and hypermanganesemia caused by mutations in SLC30A10, a manganese transporter in man. Am J Hum Genet 2012;90:457-66.
- 12. Stamelou M, Tuschl K, Chong WK, et al. Dystonia with brain manganese accumulation resulting from SLC30A10 mutations: a new treatable disorder. Mov Disord 2012;27:1317-22.
- 13. Ala A, Walker AP, Ashkan K, et al. Wilson’s disease. Lancet 2007;396:397-40.
- 14. European Association for Study of Liver. EASL Clinical Practice Guidelines: Wilson’s disease. J Hepatol 2012;56:671-85.
- 15. Roberts E, Schilsky M. Diagnosis and treatment of Wilson’s disease an update. Hepatology 2008;47:2089-111.
- 16. Machado A, Neu Chien HF, Deguti MM. Neurological manifestation of Wilson’s disease: Report of 119 cases. Mov Disord 2006;21:2192-6.
- 17. Członkowska A, Tarnacka B, Müller JC i wsp. Unified Wilson’s Disease Rating Scale – a proposal for the neurological scoring of Wilson’s disease patients. Neurol Neurochir Pol 2007;41:1-12.
- 18. Sinha S, Taly AB, Ravishankar S, et al. Wilson’s disease: Cranial MRI observations and clinical correlation. Neuroradiology 2006;48:613-21.
- 19. Ferenci P, Caca K, Loudianos G, et al. Diagnosis and phenotypic classification of Wilson disease. Liver Inter 2003;23:139-42.
- 20. Członkowska A, Gajda J, Rodo M. Effect of long-term treatment in Wilson’s disease with D-penicillamine and zinc sulphate. J Neurol 1996;243:269-73.
- 21. Członkowska A, Litwin T, Karliński M i wsp. D-penicillamine versus zinc sulfate as first-line therapy for Wilson’s disease. Eur J Neurol 2014;21:599-606.
- 22. Dhwan A, Taylor RM, Cheeseman P, et al. Wilson’s disease in children: 37-year experience and revised King’s score for liver transplantation. Liver Transpl 2005;11:441-8.
- 23. Dzieżyc K, Litwin T, Chabik G i wsp. Families with Wilson’s disease in subsequent generations: clinical and genetic analysis. Mov Disord 2014;29:1828-32.



 Dodaj do ulubionych
Dodaj do ulubionych
