



Co znajdziesz w artykule?
- Postępy w leczeniu mukowiscydozy – wprowadzenie modulatorów CFTR
- Wpływ choroby na pracę różnych układów i narządów, a także na ogólny dobrostan pacjentów
- Znaczenie kompleksowej opieki i edukacji pacjentów oraz wyzwania wynikające z wydłużenia życia chorych na mukowiscydozę
Spis treści
Mukowiscydoza (CF – cystic fibrosis), zwana najczęstszą chorobą rzadką, jest chorobą genetyczną, dziedziczoną autosomalnie recesywnie, występującą głównie u rasy kaukaskiej. Istotą patologii jest mutacja przezbłonowego kanału chlorkowego (CFTR – cystic fibrosis transmembrane conductance regulator), która prowadzi do zagęszczenia wydzielanego śluzu, a w konsekwencji do uszkodzenia wielu narządów, przede wszystkim układu oddechowego (oskrzeli, zatok obocznych nosa), ale także trzustki
(początkowo tylko zewnątrz-, ale po latach także jej wewnątrzwydzielniczej funkcji). Ponadto dochodzi do powstania niedrożności smółkowej (objaw patognomoniczny), zaburzeń wchłaniania tłuszczów i witamin w nich rozpuszczalnych oraz układu rozrodczego (nasieniowody, jajowody).
W Polsce liczba pacjentów z mukowiscydozą wynosi około 2000, z czego blisko 1/3 to osoby dorosłe. Według danych Głównego Urzędu Statystycznego z 2024 roku w Polsce jest około 10 000 specjalistów medycyny rodzinnej. To oznacza, że co 5 specjalista medycyny rodzinnej będzie miał wśród swoich stałych pacjentów chorego z mukowiscydozą, a spodziewane wydłużenie życia oraz poprawa w zakresie zdrowia prokreacyjnego mogą spowodować zwiększenie populacji chorych wymagających szczególnej opieki. Niniejszy artykuł jest próbą usystematyzowania wiedzy na temat tej choroby, opisania nowych możliwości terapeutycznych i ich konsekwencji dla życia pacjentów, ale także dla zmieniającego się obrazu schorzenia.
W Polsce od lipca 2009 roku u noworodków są wykonywane badania przesiewowe w kierunku mukowiscydozy. W 3 dobie życia pobiera się krew z pięty dziecka na bibułę w celu oznaczenia stężenia immunoreaktywnej trypsyny. W przypadku uzyskania dodatniego wyniku dzieci są kierowane na dalszą diagnostykę, która obejmuje badanie genetyczne pod kątem najczęstszych w populacji polskiej mutacji białka CFTR. U dzieci z mutacjami na obu allelach rozpoznaje się mukowiscydozę i obejmuje je specjalistyczną opieką. Dzieci z mutacją 1 allelu są kierowane także do specjalistycznego ośrodka, w którym między innymi oznacza się chlorki w pocie. Wynik tego badania determinuje ewentualne rozpoznanie mukowiscydozy przy niekonkluzywnym wyniku badań genetycznych.
Z uwagi na wysoką czułość badań przesiewowych powstaje populacja dzieci, u których nie rozpoznano mukowiscydozy mimo nieprawidłowych wyników badania przesiewowego, po przeprowadzeniu pogłębionych badań genetycznych (obejmujących większą liczbę mutacji niż badania klasyczne) lub uzyskaniu niekonkluzywnego wyniku testu potowego. Takich pacjentów określa się mianem CFSPID (Cystic Fibrosis Screen Positive, Inconclusive Diagnosis). To pacjenci, którzy powinni pozostać pod obserwacją ośrodka pediatrycznego i być monitorowani pod kątem ujawnienia się typowych dla mukowiscydozy objawów.
Badania przesiewowe obejmujące niemal całą populację noworodków w Polsce sprawiły, że choroba jest rozpoznawana na bardzo wczesnym, zwykle bezobjawowym etapie. Takie dziecko zaś od początku jest objęte opieką specjalistyczną, odpowiednią suplementacją odżywczą, ewentualną fizjoterapią bądź leczeniem mukolitycznym. Postępowanie to opóźnia nieuchronną destrukcję wielonarządową, poprawia jakość i długość życia chorych oraz umożliwia szybkie wykrywanie i leczenie powikłań.
Kolejnym krokiem milowym (żeby nie powiedzieć „rewolucyjnym”) w leczeniu mukowiscydozy było wprowadzenie w 2022 roku w ramach programu Narodowego Funduszu Zdrowia (NFZ) tzw. modulatorów białka CFTR, ogólnie nazywanych także lekami przyczynowymi. Terapia w Polsce została wdrożona zaledwie 3 lata po pierwszej rejestracji leków w USA. Nasi chorzy mają zatem dostęp do najnowocześniejszego leczenia na światowym poziomie.
Aby zrozumieć, czym są modulatory białka CFTR, należy krótko zagłębić się w genetykę samej choroby. Mutacja białka CFTR może oznaczać powstanie białka o charakterze nonsensownym (zupełnie niedziałającego, wówczas modulatory nie są skuteczne) albo takiego, które wykazuje zmniejszoną funkcję, upośledzony transport na powierzchnię błony komórkowej lub przyspieszony rozpad. Na te właśnie białka wpływają modulatory, które poprawiają ich funkcjonowanie. Spośród leków dostępne są tak zwane potencjalizatory (iwakaftor) oraz korektory (lumakaftor, tezakaftor, eleksakaftor). Dobór leków stosowanych u konkretnego pacjenta zależy od jego wieku oraz mutacji białka CFTR. Nie jest to leczenie przyczynowe w dosłownym tego słowa znaczeniu (gen nadal pozostaje uszkodzony), a niejako substytucyjne, umożliwiające lepsze funkcjonowanie nieprawidłowego białka. Wprowadzenie leków wywołało rewolucję w stanie chorych z mukowiscydozą. Pacjenci początkowo zaczęli odkrztuszać zwiększoną ilość wydzieliny, ale już po kilku dniach lub tygodniach jej objętość znacznie się zmniejszyła (niektórzy nie wykrztuszają już wcale). W związku z tym chorzy odczuwają mniejszą duszność, mniej kaszlą, mają istotnie mniej zaostrzeń. Można u nich także zaobserwować poprawę w zakresie parametrów spirometrycznych, szczególnie nasilonej pierwszosekundowej objętości wydechowej (FEV1 – forced expiratory volume in one second), której spadek jest jednym z najistotniejszych czynników predykcyjnych zgonu u tych chorych. U pacjentów widoczny jest także wzrost wskaźnika masy ciała (BMI – body mass index), nierzadko prowadzący nawet do otyłości. U chorych, u których nie doszło jeszcze do całkowitego uszkodzenia miąższu trzustki, można odnotować znamienną poprawę jej funkcji.
W związku z istotną zmianą obrazu choroby konieczne jest ponowne wyedukowanie specjalistów medycyny rodzinnej tak, aby nadal stanowili silnych partnerów w leczeniu pacjentów z mukowiscydozą. Poniżej zostaną przedstawione główne aspekty, na które należy zwrócić uwagę u chorych poddanych terapii modulatorami białka CFTR.
Układ oddechowy
Dzięki wprowadzeniu modulatorów białka CFTR zmniejszyła się częstość zaostrzeń choroby podstawowej, co (na przykładzie poznańskiego ośrodka) przedstawiono na rycinie 1 1 .
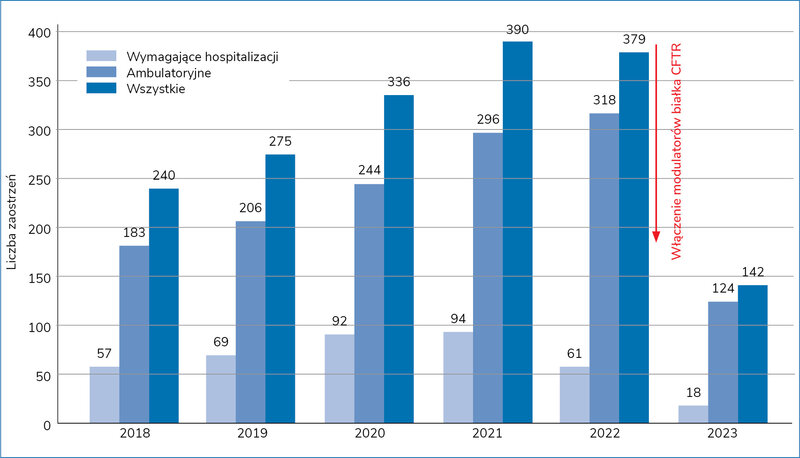
Rycina 1. Liczba zaostrzeń mukowiscydozy w ciągu roku na Oddziale Pulmonologii Uniwersyteckiego Szpitala Klinicznego w Poznaniu
Każde zaostrzenie powinno być leczone antybiotykami, zgodnie z najnowszym posiewem plwociny/wymazu z nosa i gardła/bronchofiberoskopii. W przypadku gdy wyniki ostatnich posiewów (szczególnie u pacjentów z nieproduktywnym kaszlem, mających tylko wyniki badania mikrobiologicznego z górnych dróg oddechowych) są ujemne, warto sięgnąć do ostatniego dodatniego posiewu albo zastosować antybiotykoterapię empiryczną przedstawioną w tabeli 2 3 . Pacjenci z mukowiscydozą są przewlekle skolonizowani bakteryjnie, a antybiotykoterapia nie jest w stanie doprowadzić do eradykacji drobnoustrojów. W związku z tym zakłada się, że zaostrzenie choroby zwykle wynika z namnożenia się bakterii przewlekle rezydujących w dolnych drogach oddechowych, i dlatego z powodzeniem można się odnosić do antybiogramu nawet sprzed kilku miesięcy.

Tabela 2. Antybiotykoterapia empiryczna w zaostrzeniu mukowiscydozy3
Warto sięgnąć po dawkowanie antybiotyków zamieszczone w wytycznych KOMPAS CF z 2017 roku 4 . Czas trwania antybiotykoterapii w zaostrzeniu mukowiscydozy to co najmniej 10, a w niektórych przypadkach nawet 14 dni. Dawki leków stosowanych w zaostrzeniu są wyższe niż w populacji ogólnej. Należy jednak zwrócić uwagę na interakcję niektórych leków z modulatorami białka CFTR. Dotyczy to głównie tych leków, których metabolizm odbywa się za pomocą cytochromu CYP3A4. Wówczas konieczne jest zmodyfikowanie dawkowania modulatorów zgodnie z tabelą zamieszczoną w charakterystyce produktu leczniczego.
Jeżeli nie jest możliwe przeleczenie pacjenta lekami doustnymi/wziewnymi, wymagany jest kontakt z ośrodkiem referencyjnym, który zadecyduje o ewentualnym przyjęciu chorego do szpitala na antybiotykoterapię dożylną lub o wdrożeniu finansowanego przez NFZ programu antybiotykoterapii domowej, w którym leki podaje przeszkolona pielęgniarka w domu pacjenta. Leki w ramach opisywanego programu są wydawane dla pacjenta ze szpitala za darmo, przewidziana jest także opłata za usługę pielęgniarską.
Co ważne, pacjenci ze zmniejszoną produkcją lepkiej wydzieliny niejednokrotnie zachłystują się odzyskanym życiem i zapominają o zalecanej aerozoloterapii bądź systematycznej fizjoterapii. W obserwacjach długofalowych stwierdza się niestety, że zaprzestanie fizjoterapii nawet u tych chorych, którzy nie produkują wydzieliny, negatywnie wpływa zarówno na ich stan ogólny, jak i wydolność oddechową. Konsekwencją jest obniżenie jakości życia, a w przyszłości być może wzrost ryzyka zaostrzeń. Podczas każdej wizyty trzeba więc przypominać o konieczności wykonywania rehabilitacji oddechowej, która stanowi nieodzowny sposób na wydłużenie życia pacjenta z mukowiscydozą. Jest to rola nie tylko ośrodków specjalistycznych, lecz także każdego lekarza, z którym stykają się chorzy na mukowiscydozę.
Krwioplucie
W związku z zaawansowaniem choroby podstawowej, szczególnie u pacjentów dorosłych, może się pojawiać krwioplucie, które czasem przybiera masywną postać. Do krwioplucia dochodzi zwykle w przebiegu patologii tętnic oskrzelowych (z krążenia dużego), a nie tętnic płucnych (krążenie małe). Pacjent z mukowiscydozą i krwiopluciem, który trafia na szpitalny oddział ratunkowy, oczywiście może mieć zatorowość płucną, ale najczęściej krwioplucie powstaje w wyniku malformacji tętnic oskrzelowych. Taka uwaga ma fundamentalne znaczenie w kontekście wykonywania angio-TK klatki piersiowej – w badaniu konieczne jest zobrazowanie zupełnie innych tętnic.
Postępowanie u pacjentów z krwiopluciem z tętnic oskrzelowych jest standardowe. Należy walczyć z dusznością i niedotlenieniem, koniecznie czasowo odstawić wszystkie leki wziewne (szczególnie o działaniu rozszerzającym oskrzela). Ponadto należy oznaczyć parametry krzepnięcia i wyrównywać zaburzenia; powinno się też oznaczyć grupę krwi pacjenta. Można zastosować leki uszczelniające naczynia, preparaty prokrzepliwe, można podawać dożylnie 10% NaCl lub stosować okłady z lodu na klatkę piersiową. Złotym standardem postępowania jest, po wstępnym wyrównaniu stanu chorego, wykonanie embolizacji krwawiących tętnic oskrzelowych – dostępne w kilku ośrodkach w Polsce. Jeśli nie można bezpośrednio skontaktować się z pracownią, warto poprosić o pomoc ośrodek leczenia mukowiscydozy, który ma doświadczenie w organizowaniu zarówno samego zabiegu, jak i transportu medycznego do oddalonych czasem o kilkaset kilometrów pracowni wykonujących embolizacje.
Przeszczepienie płuc
Przewlekła niewydolność oddechowa to najczęstsza postać schyłkowej fazy mukowiscydozy. Rolą ośrodka specjalistycznego jest ścisłe monitorowanie FEV1 i odpowiednio wczesne przygotowanie pacjenta do przeszczepienia płuc. Po wprowadzeniu modulatorów białka CFTR konieczność wykonywania zabiegów przeszczepienia płuc radykalnie zmalała, co pokazuje raport Cystic Fibrosis Foundation z 2024 roku (ryc. 2) 5 .
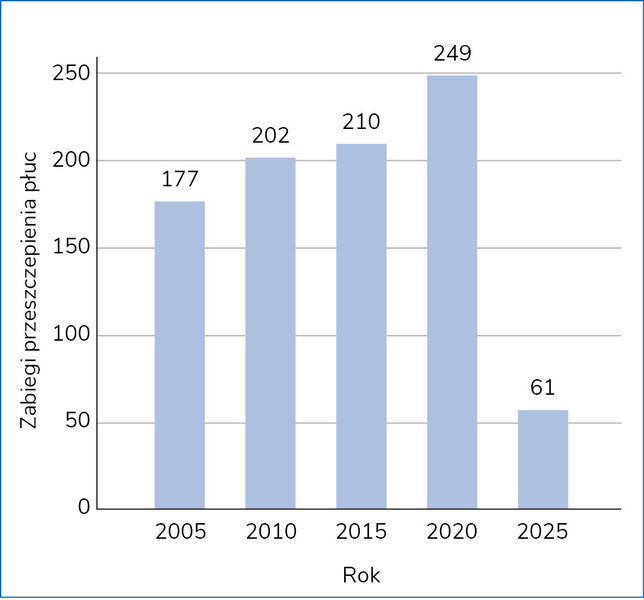
Rycina 2. Liczba przeszczepów płuc w latach 2004-2025 według Cystic Fibrosis Foundation Patient Registry Highlights5
Wydaje się jednak, że choć modulatory (szczególnie wprowadzone dopiero u dorosłych chorych z dokonanymi zniszczeniami układu oddechowego) opóźnią konieczność przeszczepu, nie sprawią, że transplantacje płuc nie będą wymagane. Stosowanie tej terapii przesunie tylko konieczność wykonania przeszczepienia na znacznie późniejszy wiek pacjenta. Dlatego też pacjenci powinni szczególnie dbać m.in. o stan uzębienia i leczyć ogniska zapalne, tak aby potrzeba wizyty u stomatologa nie opóźniała możliwości transplantacji. U chorych po przeszczepieniu płuc należy pamiętać o systematycznym pomiarze FEV1 (chorzy mają zwykle możliwość przeprowadzania domowych pomiarów) i wczesnej reakcji na spadek tego parametru. Obligatoryjne są agresywne leczenie infekcji oraz okresowa weryfikacja flory zasiedlającej drogi oddechowe. W przypadku niektórych leków (np. przeciwgrzybiczych) należy pamiętać o weryfikowaniu dawek leków immunosupresyjnych.
Stan po przeszczepieniu płuc aktualnie stanowi przeciwwskazanie do stosowania modulatorów białka CFTR.
Układ pokarmowy
Pacjenci z mukowiscydozą zwykle od dzieciństwa zmagają się z tłuszczowymi biegunkami i zaburzeniami wchłaniania tłuszczów. Wynika to między innymi z upośledzenia wydzielania do jelit soku trzustkowego i nieprawidłowego trawienia tłuszczu, uniemożliwiającego jego wchłanianie. Zatem chorzy, aby utrzymać optymalną masę ciała, znacznie zwiększali podaż kaloryczną, ponieważ substytucja enzymów trzustkowych nie była w stanie w pełni wyrównać zaburzeń wchłaniania. Jeszcze niedawno około 20% chorych z mukowiscydozą było niedożywionych 6 . Aktualnie, po wdrożeniu leczenia modulatorami białka CFTR, szczególnie u tych pacjentów, u których nie doszło jeszcze do całkowitej degradacji trzustki, występuje znacznie lepsze wchłanianie jelitowe, co wraz z redukcją wydatku kalorycznego związanego z kaszlem, oddechem przy większej obturacji itd., poprawą węchu i smaku, a także zwiększeniem apetytu skutkuje zwiększeniem masy ciała 7 .
Obecnie wzrasta liczba chorych z nadwagą, a nawet otyłością. Wynika to z konieczności szybkiego, niemal całkowitego przestawienia celów dietetycznych, rodzajów posiłków lub liczby spożywanych kalorii. Niestety zwłaszcza u osób w wieku nastoletnim widoczny jest niepokojący trend: nastolatki z obawy przed przytyciem pomijają dawki leków albo czasowo je odstawiają, tak aby ponownie doprowadzić do zaburzeń wchłaniania i dzięki temu utrzymać wymarzoną sylwetkę. Ważne są zatem edukacja dotycząca całej terapii mukowiscydozy, reedukacja dotycząca diety oraz ponowne proponowanie fizjoterapii jako jednego z czynników warunkujących utrzymanie zdrowej sylwetki z optymalnym BMI. Każdorazowe ważenie pacjentów podczas wizyt lekarskich i w miarę możliwości poruszanie problemu diety, zanim dojdzie do nadwagi, stanowią istotne działania w profilaktyce zdrowotnej u pacjentów z mukowiscydozą.
U chorych z niewydolnością zewnątrzwydzielniczą trzustki (absolutna większość pacjentów dorosłych) konieczne jest utrzymanie substytucji pankreatyny w ilości odpowiadającej zawartości tłuszczu w posiłkach. Pacjenci wiedzą, jak przeliczać dawki pankreatyny na ilość tłuszczu. Dlatego mimo że czasem dawki stosowanej pankreatyny wydają się absurdalne (do kilkunastu kapsułek na dobę), należy zaufać pacjentowi i zaopatrzyć go w odpowiednią ilość środka przepisywanego na receptę. Nadal obowiązuje suplementacja witamin rozpuszczalnych w tłuszczach. Ośrodki leczenia mukowiscydozy okresowo sprawdzają ich stężenia we krwi i modyfikują dawkę suplementacji. Należy pamiętać, że u chorych wciąż istnieje zwiększone ryzyko osteoporozy, dlatego konieczne są okresowa ocena densytometryczna i wczesne włączanie klasycznego leczenia osteoporozy.
W związku z niedrożnością drobnych przewodów wewnątrzwątrobowych, leczeniem hepatotoksycznym oraz ryzykiem zakażenia wirusem WZW typu B i C związanym z licznymi hospitalizacjami pacjenci z mukowiscydozą są narażeni na rozwój hepatopatii, a czasem nawet marskości wątroby. U części pacjentów zachodzi konieczność przeszczepienia wątroby. W opiece nad pacjentami z hepatopatią należy pamiętać o ich systematycznym monitorowaniu pod kątem powikłań, między innymi pojawienia się żylaków przełyku (umożliwia to ich opaskowanie, zanim dojdzie do istotnego krwawienia).
Cukrzyca typu 3
U pacjentów z mukowiscydozą niejednokrotnie po uszkodzeniu zewnątrzwydzielniczej funkcji trzustki obserwuje się zaburzenie części wewnątrzwydzielniczej i powstanie cukrzycy typu 3. W związku z upośledzeniem produkcji insuliny konieczna jest insulinoterapia; aktualnie nie ma wskazań do stosowania leków działających na mechanizm insulinooporności. Wydaje się, że na skutek wzrostu masy ciała u chorych z cukrzycą, wczesnego włączania leczenia przyczynowego i opóźnienia lub nawet zniesienia ryzyka uszkodzenia trzustki za kilkanaście lat profil diabetologiczny pacjentów z mukowiscydozą może się diametralnie zmienić i tak jak w populacji ogólnej może zacząć się pojawiać cukrzyca typu 2 związana z insulinoopornością. Na razie pozostaje to kwestią przyszłości, jednak diabetolodzy zaangażowani w leczenie cukrzycy związanej z mukowiscydozą zachowują pod tym względem zwiększoną czujność.
Zdrowie prokreacyjne
Dobre samopoczucie pacjentów, lepsze odżywienie i większa wydolność wysiłkowa sprawiają, że pacjentki częściej decydują się na ciążę, ponieważ wiedzą, że ich stan zdrowia aktualnie na to pozwala. To oczywiście świetna wiadomość. Według raportu Cystic Fibrosis Foundation 5 liczba ciąż wśród pacjentek z mukowiscydozą w USA wzrosła z 310 w 2018 roku do 655 w 2024 roku. Niestety wśród części pacjentów pokutuje przekonanie, że skoro ich choroba wiąże się z upośledzeniem płodności, nie muszą myśleć o ciąży jako konsekwencji współżycia. Szacuje się, że około 43% aktywnych seksualnie osób z mukowiscydozą stosuje antykoncepcję, w porównaniu z około 76% populacji ogólnej 8 . Te liczby nie wydają się wynikać z odmiennego podejścia do planowania rodziny, tylko z niewiedzy pacjentów. W związku z tym konieczna jest systematyczna edukacja seksualna. Ważne jest podejmowanie problemu zdrowia prokreacyjnego podczas wizyt lekarskich. Tylko dobrze wyedukowany pacjent będzie w stanie świadomie decydować się na macierzyństwo bądź ojcostwo. Będzie to też szansa dla medyków na dobre przygotowanie zdrowotne pacjentek do zajścia w ciążę. Istnieje także możliwość przebadania partnerów pod kątem nosicielstwa mutacji białka CFTR w ramach porady genetycznej przed planowaną ciążą. W przypadku wykrycia mutacji ryzyko urodzenia dziecka z mukowiscydozą wzrasta do 50%.
Z pacjentkami planującymi ciążę, a przyjmującymi leki przyczynowe należy porozmawiać o ich stosowaniu w tym okresie. Dotychczas obserwowane pacjentki i ich dzieci nie wykazywały zwiększonej częstości powikłań w związku ze stosowaniem leków przyczynowych. Wręcz przeciwnie: pacjentki przechodzą przez ciążę lepiej, z mniejszą dusznością i częstością zaostrzeń. Zalecenia European Cystic Fibrosis Society (ECFS) mówią o pozostawieniu leków przyczynowych u pacjentek przez całą ciążę lub ewentualnym wstrzymaniu terapii w pierwszym trymestrze. Niemniej ponieważ w charakterystyce produktów leczniczych nie ma stosownych zapisów, każdorazowo należy porozmawiać z pacjentką o ewentualnym utrzymaniu leków przyczynowych w ciąży i uzyskać jej zgodę na taką terapię.
Dobrostan socjalny
Wraz z poprawą stanu zdrowia pacjentów z mukowiscydozą zaczynają się pojawiać problemy o charakterze socjalnym. Po wprowadzeniu modulatorów białka CFTR stan zdrowia chorych poprawił się na tyle, że mogli na przykład po raz pierwszy pójść do szkoły, zamiast korzystać z indywidualnego nauczania w domu. Emocje związane z wejściem do świata rówieśniczego, diametralna zmiana sposobu nauczania, pojawienie się rywalizacji – to zupełnie nowe wyzwania, dotyczą chorych, którzy nierzadko dopiero w wieku nastoletnim zaczynają swoją pierwszą w życiu przygodę z prawdziwą szkołą.
Ponadto poprawa wydolności fizycznej, zmniejszenie czasu poświęcanego na aerozoloterapię i rehabilitację sprawiają, że chorzy mogą nie otrzymywać już części dotychczasowych świadczeń socjalnych. Nierzadko pacjenci nie mają wykształcenia umożliwiającego im pracę umysłową, a do pracy fizycznej zwyczajnie są niezdolni. Dotychczasowy, często dramatyczny przebieg choroby, liczne zaostrzenia i hospitalizacje znacznie utrudniały ich edukację. To wielki problem wymagający rozwagi ze strony lekarzy orzeczników i wzięcia pod uwagę całokształtu choroby, także z czasu przed włączeniem leczenia modulatorami białka CFTR, tak by nie skrzywdzić pacjentów poprzez ograniczenie im dostępu do środków niezbędnych do życia oraz dalszej rehabilitacji.
Onkologia w mukowiscydozie
W związku z dotychczasową krótką przeżywalnością pacjentów z mukowiscydozą dane onkologiczne odnoszące się do tej grupy były ograniczone. Wraz z rozwojem opieki i wydłużaniem czasu przeżycia okazało się jednak, że pacjenci z mutacją białka CFTR mają zwiększone ryzyko zapadalności na niektóre nowotwory. Dane wskazują przede wszystkim na: raka przewodu pokarmowego (ryzyko zwiększone 3,5-krotnie), raka jąder (ryzyko o 1,7 razy wyższe) i białaczkę limfatyczną (ryzyko 2-krotnie wyższe) 9 .
Te dane zaowocowały powstaniem wstępnych zaleceń dotyczących np. badań przesiewowych w kierunku raka jelita grubego. U pacjentów z mukowiscydozą kolonoskopia powinna być wykonywana od 40 roku życia co 5 lat albo co 3 lata, o ile w jelicie stwierdzono polipy. Na marginesie warto dodać, że mutacja w białku CFTR sprzyja powstawaniu polipów przewodu pokarmowego. Zalecenia są jeszcze bardziej restrykcyjne u osób po przeszczepieniu narządów: kolonoskopia powinna się odbywać z wyżej opisaną częstością od 30 roku życia lub gdy miną co najmniej 2 lata od transplantacji. Niestety, gdy porównano grupę pacjentów po transplantacji w przebiegu mukowiscydozy z pacjentami po transplantacji z innych powodów, ryzyko u chorych z mukowiscydozą było znamiennie wyższe niż w pozostałych grupach.
Szczególnie ważne okazuje się także zachęcanie mężczyzn do samobadania jąder lub przeprowadzanie takiego badania podczas wizyty lekarskiej. Dzięki temu wzrosną szanse na wykrycie nowotworu w stadium choroby miejscowej.
Trudno wprowadzić badania przesiewowe w kierunku nowotworów hematologicznych. Niemniej w przypadku utraty masy ciała, nawracającej gorączki bądź nocnych potów warto pamiętać, że opisane objawy mogą się wiązać nie tylko z zaostrzeniem choroby płuc albo zakażeniem, lecz także z nowotworem hematologicznym, który wymaga pilnej, agresywnej diagnostyki.
Pozostałe choroby cywilizacyjne
W związku z wydłużeniem spodziewanej długości życia u pacjentów z mukowiscydozą należy się spodziewać, że w tej grupie będą występować udary mózgu, zawały serca bądź inne choroby związane z miażdżycą. Aktualnie dane dotyczące ryzyka tych chorób u pacjentów z mukowiscydozą są skromne. Wiemy natomiast, że pacjenci z mukowiscydozą coraz częściej będą utrzymywać prawidłową (a czasem nawet zwiększoną) masę ciała, że jednym z powikłań choroby jest cukrzyca, że chorzy często mają nieprawidłowy profil lipidowy i że przewlekły stan zapalny w ich organizmach jest samodzielnym czynnikiem ryzyka rozwoju miażdżycy. Dlatego najprawdopodobniej wraz z wydłużeniem życia będzie u nich rosło ryzyko chorób układu sercowo-naczyniowego. W diagnostyce różnicowej warto więc pamiętać także o tych chorobach.
Działania niepożądane
Wraz z pojawieniem się nowych grup leków w terapii mukowiscydozy zaczęły występować ich działania niepożądane, na które lekarze rodzinni powinni zwracać szczególną uwagę. Są to przede wszystkim uszkodzenia wątroby (z nierzadko bardzo wysokimi aktywnościami transaminaz we krwi), zmiany skórne o charakterze wysypki lub bezsenność. Należy pytać pacjentów o ich samopoczucie emocjonalne oraz problemy ze snem podczas każdej wizyty lekarskiej, tak aby niezwłocznie zmniejszyć niepożądane skutki leczenia.
Modulatory białka CFTR z jednej strony mogą wykazywać działanie hepatotoksyczne, z drugiej natomiast ich niemały wpływ na poprawę ogólnego stanu zdrowia sprawia, że z ostrożnością mogą być stosowane u pacjentów z wyrównaną metabolicznie marskością wątroby, a także u tych po przeszczepieniu tego narządu. W tych grupach chorych obowiązuje ścisłe monitorowanie czynności wątroby.
Należy pamiętać także o wspomnianym już szlaku metabolizowania modulatorów białka CFTR i dostosowywaniu dawki modulatorów w przypadku konieczności przyjmowania leków wpływających na aktywność cytochromu CYP3A4. Charakterystyka omawianych produktów leczniczych jest zaopatrzona w tabele dokładnie opisujące sposób i czas trwania redukcji dawki leków. Warto mieć to na uwadze.
Skąd czerpać wiedzę?
W związku z dynamiczną sytuacją kliniczną pacjentów z mukowiscydozą warto sięgać po sprawdzone źródła wiedzy. Na stronach Polskiego Towarzystwa Mukowiscydozy, w zakładce „Dla profesjonalistów” można znaleźć systematycznie aktualizowane wytyczne zarówno dla lekarzy, jak i fizjoterapeutów lub psychologów. Ponadto warto przestudiować następujące opracowania:
- Mielus M, Walicka-Serzysko K, Sands D. Rozpoznawanie i leczenie mukowiscydozy. Podsumowanie wytycznych European Cystic Fibrosis Society 2018. Med Prakt 2019;6:e1-e17 – polskie opracowanie wytycznych ECFS; dokładnie omówiono w nim wszystkie medyczne aspekty związane z diagnostyką i leczeniem mukowiscydozy
- Castellani C, Duff AJA, Bell SC, et al. ECFS best practice guidelines: the 2018 revision. J Cyst Fibros 2018;17(2):153-78 – w dokumencie podsumowano najważniejsze zmiany w postępowaniu u pacjentów z mukowiscydozą po wprowadzeniu modulatorów białka CFTR
- Sands D, Walicka-Serzysko K, Doniec Z i wsp. ReKOMendacje PostępowAnia w mukowiScydozie (cystic fibrosis; CF) dla lekarzy Podstawowej Opieki Zdrowotnej – KOMPAS CF – część 1. Pediatria Polska 2017:92(4):431-45. https://doi.org/10.1016/j.pepo.2017.04.006.
Podsumowanie
Mukowiscydoza pozostaje trudną, wielonarządową, nieuleczalną chorobą, która w sposób istotny wpływa na życie pacjenta i jego bliskich. Coraz nowocześniejsze terapie sprawiają jednak, że jej obraz się zmienia. Pacjenci żyją dłużej i w lepszym komforcie, choć taka zmiana rodzi nowe wyzwania zarówno dla pracowników opieki zdrowotnej, jak i samych chorych. Dopiero dobra współpraca wszystkich ogniw – od poradni lekarza rodzinnego po specjalistyczne ośrodki leczenia mukowiscydozy, od badań przesiewowych noworodków po przeszczepienia płuc – jest w stanie maksymalnie wydłużyć długość i jakość życia pacjentów z mukowiscydozą.
Abstract
Cystic fibrosis: A (not)rare disease
Cystic fibrosis (CF) is the most common rare genetic disease among Caucasians, caused by mutations in the CFTR gene, leading to productive viscous secretions and progressive multi-organ damage. In Poland, there are about 2,000 patients living with CF, one-third of whom are adults. The newborn screening program in place since 2009 has enabled providing an early diagnosis and specialist care from the infancy period. The introduction of CFTR modulators, which in Poland have been listed as reimbursable drugs since 2022, has revolutionized CF treatment. These drugs, including potentiators and correctors, improve the function of defective CFTR protein, leading to fewer pulmonary exacerbations, improved lung function, nutritional status and overall quality of life.
This therapeutic shift changes the disease trajectory and creates new challenges for family physicians who are often the first point of contact. Clinical aspects requiring particular attention include: diagnosis and treatment of exacerbations, management of hemoptysis and chronic infection, transplant-related care, nutritional issues with a growing prevalence of overweight, CF-related diabetes, liver diseases, bone health, and emerging risks of malignancy. In addition, reproductive health, psychosocial adaptation and socioeconomic challenges have become increasingly relevant as patients live longer and healthier lives. Physicians must also monitor patients for adverse effects of CFTR modulators, particularly hepatotoxicity and CYP3A4-mediated drug interactions.
The evolving natural history of CF highlights the importance of continuous education for family physicians and close collaboration with specialist centers. Only a comprehensive, multidisciplinary approach, from newborn screening to advanced therapies and transplant care, can help to maximize both longevity and quality of life for patients with CF.
- 1. Winiarska HM, Springer D, Wojtaś F, et al. CFTR Modulators Therapy Efficacy in Reducing Cystic Fibrosis (CF) Exacerbation and Improving Selected Spirometry Parameters: A Real-Life Study in a Single-Centre Polish Population. J Clin Med 2024;13(15):4491. doi: 10.3390/jcm13154491
- 2. VanDevanter DR, Hamblett NM, Simon N, et al. Evaluating assumptions of definition-based pulmonary exacerbation endpoints in cystic fibrosis clinical trials. J Cyst Fibros 2021;20(1):39-45
- 3. Szczeklik A, Gajewski P (red.). Interna Szczeklika. Kraków: Medycyna Praktyczna, 2025
- 4. Sands D, Walicka-Serzysko K, Doniec Z i wsp. ReKOMendacje PostępowAnia w mukowiScydozie (cystic fibrosis; CF) dla lekarzy Podstawowej Opieki Zdrowotnej – KOMPAS CF – część 1. Pediatria Polska 2017:92(4):431-45. https://doi.org/10.1016/j.pepo.2017.04.006
- 5. 2024 Cystic Fibrosis Foundation Patient Registry Highlights. Bethesda, Maryland: Cystic Fibrosis Foundation, 2025
- 6. Solís-García M, García-Clemente MM, Madrid-Carbajal CJ, et al. Is Obesity a Problem in New Cystic Fibrosis Treatments? Nutrients 2024;16(18):3103. doi: 10.3390/nu16183103
- 7. Leonard A, Bailey J, Bruce A, et al. Nutritional considerations for a new era: A CF foundation position paper. J Cyst Fibros 2023;22(5):788-95. doi: 10.1016/j.jcf.2023.05.010
- 8. Elson EC, Imburgia T, Lonabaugh K, et al. Pharmacologic contraception methods for people with cystic fibrosis: A practical review for clinicians. J Cyst Fibros 2024;23(4):653-7. doi: 10.1016/j.jcf.2024.01.003
- 9. Indra R, Černá V. The relationship between cancer risk and cystic fibrosis: the role of CFTR in cell growth and cancer development. RSC Med Chem 2025;16(8):3416-28. doi: 10.1039/d5md00203f
Następny artykuł:


 Dodaj do ulubionych
Dodaj do ulubionych
