


Spis treści
Rozwarstwienie siatkówki związane z chromosomem X jest jedną z częstszych genetycznych chorób siatkówki, w których dochodzi do zwyrodnienia plamki żółtej. Występuje prawie wyłącznie u mężczyzn, a objawy pojawiają się już w pierwszej dekadzie życia. W artykule omówiono szczegółowo patofizjologię schorzenia, objawy chorobowe, diagnostykę oraz zwrócono uwagę na leczenie, w tym terapię genową.
Wprowadzenie
Rozwarstwienie siatkówki związane z chromosomem X (XLRS – X-linked retinoschisis) zostało
po raz pierwszy rozpoznane przez austriackiego okulistę Josefa Hassa u dwóch chorych braci w 1898 r. 1 W zależności od badanej populacji częstość występowania waha się w granicach od 1:5000 do 1:25 000 urodzonych chłopców. Jest to wrodzone zaburzenie siatkówki zaliczane do witreoretinopatii, występujące prawie wyłącznie u mężczyzn. Nosicielkami są kobiety, na ogół bez zmian na dnie oka, chociaż w piśmiennictwie znajdują się pojedyncze doniesienia o heterozygotach, u których pojawiły się objawy kliniczne. 2, 3
Rozwarstwienie siatkówki związane z chromosomem X jest spowodowane mutacjami w genie RS1, który znajduje się na chromosomie Xp22.2. 4 Zidentyfikowano ponad 190 mutacji w obrębie tego genu związanych z tą chorobą. Gen ten obejmuje 32,4 kb genomowego DNA i koduje 224-aminokwasowe zewnątrzkomórkowe białko, retinoschizynę. Białko to – o masie atomowej 24-kDa – zawiera domenę diskoidyny, która znajduje się w siatkówce i szyszynce. 5 Uważa się, że domena ta bierze udział w interakcjach międzykomórkowych na powierzchni błony i jest kluczowa dla funkcjonowania białka RS1. 6, 7, 8 Retinoschizyna wydzielana jest z komórek fotoreceptorów zewnętrznych warstw siatkówki i komórek dwubiegunowych znajdujących się w warstwach wewnętrznych siatkówki neurosensorycznej. Jej rola polega na stabilizacji komórkowej synaps fotoreceptorowych dwubiegunowych oraz organizacji komórkowej siatkówki. 9, 10 Białko RS1 jest również powiązane z sodowo-potasową (Na+/K+) ATP-azą znajdującą się w wewnętrznych segmentach błon fotoreceptorów. 11 W wyniku zaburzenia białka retinoschizyny i równowagi jonowej pompy sodowo-potasowej dochodzi do zaburzenia utrzymania integralności fotoreceptorów skutkującego gromadzeniem się płynu zewnątrzkomórkowego w przestrzeniach siatkówki, które prowadzi do rozwarstwienia. 12
Obraz kliniczny choroby
Najczęstszym objawem klinicznym choroby jest pogorszenie ostrości wzroku. Schorzenie ujawnia się głównie w pierwszej i drugiej dekadzie życia, a objawia przede wszystkim trudnościami w czytaniu. Pacjenci mogą również zgłaszać objawy dyschromatopsji, najczęściej w zakresie barw czerwonej i zielonej. 13 Rzadziej choroba występuje w wieku niemowlęcym, kiedy objawia się najczęściej w postaci zeza towarzyszącego, oczopląsu, nadwzroczności osiowej, którym towarzyszą obwodowe rozwarstwienia siatkówki oraz często krwotok do ciała szklistego. 14
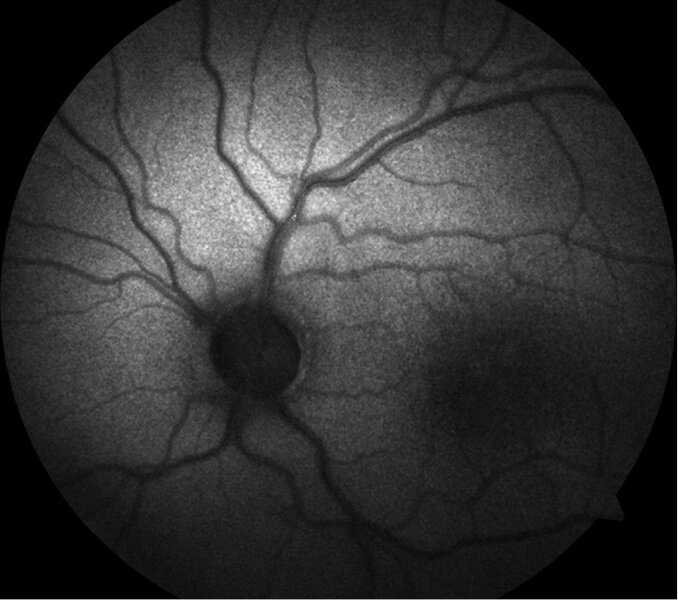
Charakterystyczną cechą fenotypową u pacjentów z rozpoznanym XLRS w badaniu dna oka jest rozwarstwienie dołka plamki. Objaw ten występuje u około 98-100% pacjentów. 15 Rozwarstwienie to ma postać małych torbieli zlokalizowanych w dołku plamki i układa się w gwiaździsty wzór, który w badaniu oftalmoskopowym może tworzyć obraz koła szprychowego (ryc. 1).
Obwodowe rozwarstwienie siatkówki występuje u około 50% pacjentów i najczęściej lokalizuje się w kwadrancie dolnoskroniowym. 16 Inne obserwowane zmiany, takie jak podsiatkówkowe liniowe zwłóknienia, „welony szklistkowe”, pigmentacje, obszary plamistej siatkówki oraz osłabienie osłonek naczyniowych, często pojawiają się w obwodowej siatkówce.
Chorobę można podzielić na 4 stadia:
- typ 1 – rozwarstwienie dołka, bez cech rozwarstwienia siatkówki; w badaniu optycznej koherentnej tomografii (OCT – optical coherence tomography) rozwarstwienie nie wykracza poza obszar widoczny w badaniu oftalmoskopowym
- typ 2 – rozwarstwienie dołka i rozwarstwienie warstwowe widoczne w OCT i wykraczające poza obszar widoczny w badaniu dna oka, bez obwodowego rozwarstwienia siatkówki
- typ 3 – rozwarstwienie dołka, warstwowe oraz obwodowe rozwarstwienie siatkówki
- typ 4 – rozwarstwienie dołka oraz obwodowe siatkówki.
Najczęściej spotykany jest typ 3, średnio u około 71% pacjentów. 17
Przebieg choroby, rokowanie i powikłania młodzieńczego rozwarstwienia siatkówki związanego z chromosomem X
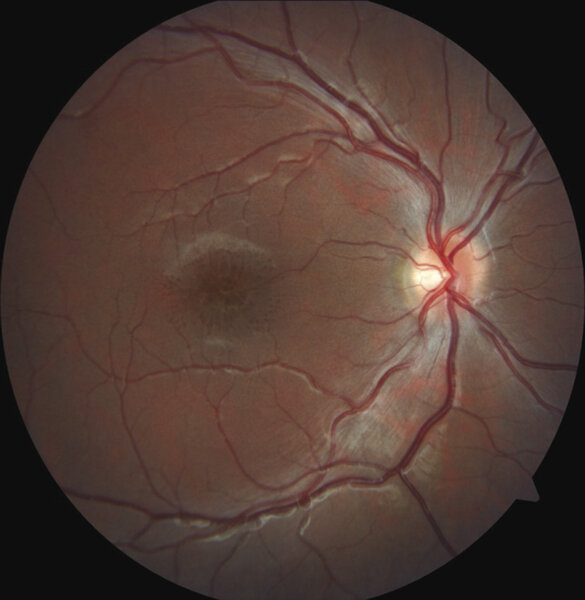
Rycina 1. Dno oka u pacjenta z rozwarstwieniem siatkówki związanym z chromosomem X. W obrębie dołka plamki widoczne promieniste fałdy siatkówki tworzące obraz kół szprychowych. Wszystkie zamieszczone zdjęcia pochodzą z materiałów własnych
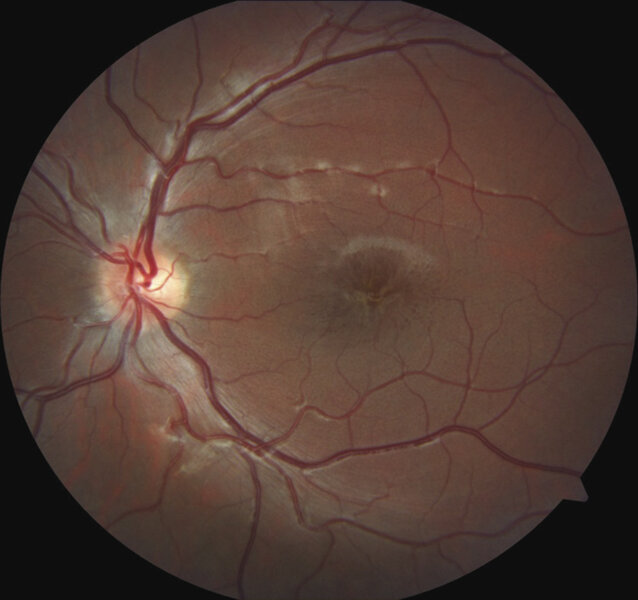
Rycina 1. Dno oka u pacjenta z rozwarstwieniem siatkówki związanym z chromosomem X. W obrębie dołka plamki widoczne promieniste fałdy siatkówki tworzące obraz kół szprychowych. Wszystkie zamieszczone zdjęcia pochodzą z materiałów własnych
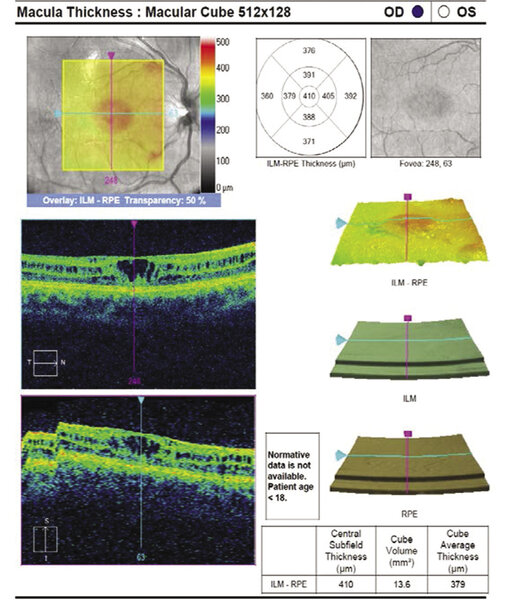
Rycina 2. Typowa makulopatia (rozwarstwienie dołka) w przebiegu XLRS. Widoczne cystowate przestrzenie umiejscowione najczęściej w warstwie jądrzastej wewnętrznej i zewnętrznej warstwie splotowatej, ponadto uwagę zwracają zdezorganizowanie struktury dołeczka i pogrubienie centralnej siatkówki
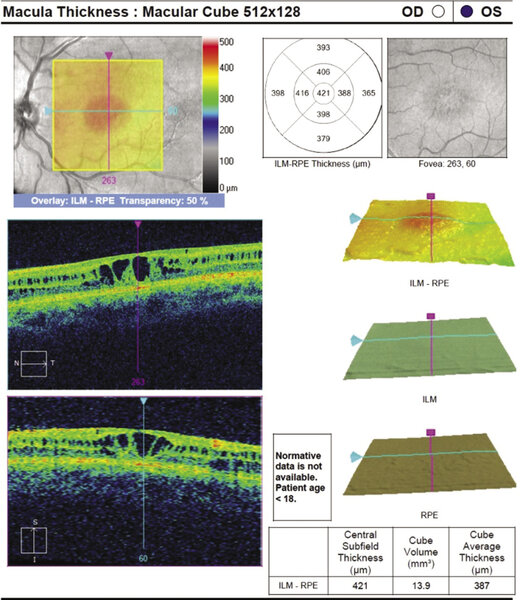
Rycina 2. Typowa makulopatia (rozwarstwienie dołka) w przebiegu XLRS. Widoczne cystowate przestrzenie umiejscowione najczęściej w warstwie jądrzastej wewnętrznej i zewnętrznej warstwie splotowatej, ponadto uwagę zwracają zdezorganizowanie struktury dołeczka i pogrubienie centralnej siatkówki
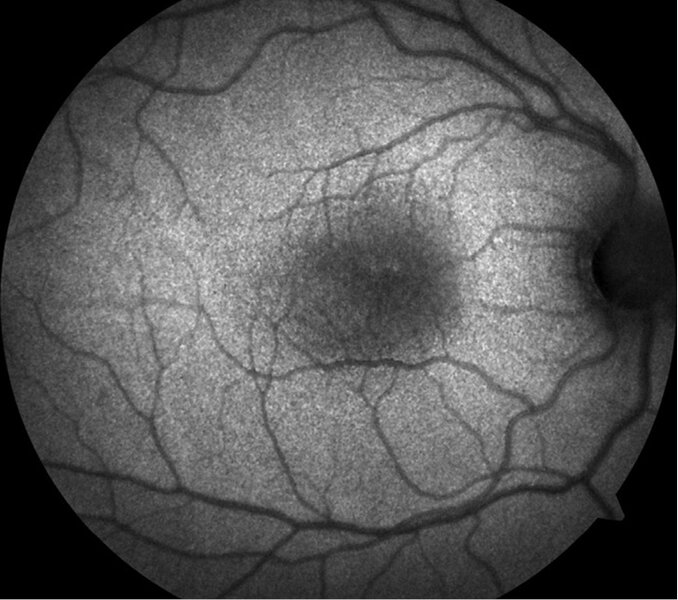
Rycina 3. Hipoautofluorescencja w obrębie dołka plamki
Rycina 3. Hipoautofluorescencja w obrębie dołka plamki
U 5-20% pacjentów może dojść do odwarstwienia siatkówki (trakcyjnego lub przedarciowego). 19 Krwotok do ciała szklistego pojawia się u około 1/3 pacjentów. 20 Są to najpoważniejsze powikłania XLRS, podczas których dochodzi do znacznej utraty ostrości wzroku. Do innych, rzadziej występujących powikłań należy zaliczyć: krwawienie w obrębie obszarów rozwarstwienia siatkówki, 21 rozszczepienie warstw siatkówki obejmujące plamkę żółtą, jaskrę neowaskularną, 22 trakcje szklistkowo-siatkówkowe pociągające obszar plamki żółtej, 23, 24 atrofię nerwu wzrokowego, 25 nowotworzenie naczyń, wysięki podsiatkówkowe oraz pourazowe pęknięcie rozwarstwienia w dołku.
Diagnostyka różnicowa
Diagnostyka różnicowa rozwarstwienia siatkówki związanego z chromosomem X powinna obejmować schorzenia takie jak rodzinna wysiękowa witreoretinopatia (zespół Criswicka-Schepensa), zespół wzmożonej aktywności czopków krótkofalowych (enhanced S-cone syndrome) i zespół Goldmanna-Favre’a (degeneracja szklistkowo-siatkówkowa), spowodowane mutacją w genie NR2E3, w których to również może dojść do rozwarstwienia plamki. Jednym z kluczowych badań niezbędnych w diagnostyce różnicowej pozostaje elektroretinogram (ERG), który różni się od typowego zapisu charakterystycznego dla XLRS (znacznie zredukowane fale a i b ze zmienionym czasem) i pozwala na różnicowanie tych jednostek chorobowych. 26, 27, 28
Innymi chorobami, które należy rozważyć podczas diagnostyki różnicowej, są: nabyte zwyrodnieniowe rozwarstwienie siatkówki, torbielowaty obrzęk plamki, odwarstwienie siatkówki; objawy mogą również naśladować chorobę Ealesa i chorobę Coatsa. Ponadto typowy zapis ERG u pacjentów z XLRS (ujemna lub zredukowana fala b) może dotyczyć także osób z wrodzoną ślepotą nocną.
Badania dodatkowe
Typowy obraz kliniczny choroby wraz z wywiadem rodzinnym i wynikami badań elektrofizjologicznych sprawiają, że ustalenie wstępnego rozpoznania najczęściej jest prawidłowe.
Rozpoznanie ostateczne można potwierdzić badaniem mutacji genu RS1. Dodatkowymi badaniami, które mogą być użyteczne w dokumentowaniu progresji choroby, są między innymi OCT, autofluorescencja, angiografia fluoresceinowa.
Badanie elektrofizjologiczne, czyli ERG, klasycznie wykazuje zmniejszenie amplitudy fali b z relatywnie zachowaną amplitudą fali a, chociaż w niektórych przypadkach można zaobserwować zachowaną amplitudę fali b. 29 Zmiana współczynnika fali b/a (tzw. ujemna fala b) występuje u około 50% pacjentów, a relatywnie prawidłowy wynik ERG nie wyklucza rozpoznania XLRS. 30, 31, 32 Multifokalny ERG (mfERG) może również wykazać zmniejszenie amplitudy fali i wydłużenie czasu kulminacji w obrębie centrum plamki.
OCT najczęściej wykazuje cystowate przestrzenie występujące w warstwie jądrzastej wewnętrznej i zewnętrznej warstwie splotowatej z cechami zdezorganizowania dołka plamki (ryc. 2).
Autofluorescencja może uwidocznić centralną hipoautofluorescencję w obrębie plamki żółtej (ryc. 3).
Angiografia fluoresceinowa, najczęściej w towarzyszącej makulopatii, obrazuje ubytki okienkowe, ale bez cech przecieku, w przeciwieństwie do torbielowatego obrzęku plamki.
Postępowanie
W przeprowadzonych u pacjentów z XLRS badaniach wykazano, że miejscowe podawanie 2% dorzolamidu (inhibitora anhydrazy węglanowej) wyraźnie zmniejsza grubość siatkówki w dołku i wiąże się z poprawą ostrości wzroku. Nie wszyscy pacjenci jednak odpowiadają na to leczenie, a u niektórych poprawa może być widoczna dopiero po kilku miesiącach. 33, 34, 35 Ponadto zaobserwowano, że ogólne podanie dorzolamidu w dawce 500 mg na dobę również może korzystnie wpłynąć na poprawę strukturalną rejonu plamki i ostrości widzenia. 36
Interwencja chirurgiczna z zastosowaniem witrektomii najczęściej jest zarezerwowana dla pacjentów z ciężkimi powikłaniami, takimi jak odwarstwienie siatkówki i krwotok do ciała szklistego. 37
Profilaktyczne leczenie rozwarstwienia za pomocą laseroterapii lub chirurgii witreoretinalnej nie jest zalecane z powodu możliwych długotrwałych ciężkich powikłań, na przykład odwarstwienia siatkówki. 19, 38
Terapia genowa
W badaniach przeprowadzonych na myszach z XLRS wykazano, że obecna w siatkówce retinoschizyna ulega ekspresji podczas wczesnego rozwoju i utrzymuje się przez całe życie, zastąpienie genów wydaje się zatem obiecującym sposobem leczenia pacjentów z rozwarstwieniem siatkówki. Dowiedziono, że wstrzyknięcie do ciała szklistego wektora genu RS1 powoduje, iż retinaza ulega ekspresji we wszystkich warstwach siatkówki, co przekłada się na poprawę amplitudy fali b w ERG. Nadal jednak trwają badania nad dostosowaniem odpowiedniej dawki genu, wyborem wektora oraz bezpieczeństwem terapii. 39, 40
Podsumowanie
Mimo dokładnego poznania patofizjologii rozwarstwienia siatkówki związanego z chromosomem X oraz trwających zaawansowanych badań genetycznych w dalszym ciągu medycyna nie umożliwia leczenia przyczynowego. Najnowsze doniesienia naukowe dają jednak duże nadzieje związane z terapią genową, która może okazać się bardzo skuteczną metodą leczenia.
Abstract
X-linked retinoschisis
X-linked retinoschisis is a common genetic diseases of the retina, which involves macular degeneration. It affects almost exclusively men and the symptoms become apparent before the end of the first decade of life. This paper details the pathophysiology, symptoms and diagnosis of X-linked retinoschisis, including a brief discussion of the existing treatment options, e.g. gene therapy.
- 1. Haas J. Ueber das zusammenvorkommen von veranderungen der retina und choroidea. Arch Augenheikd 1898;37:343-8.
- 2. Gieser EP, Falls HF. Hereditary retinoschisis. Am J Ophthalmol 1961;51:1193-200.
- 3. Wu G, Cotlier E, Braudie S. A carrier state of X-linked juvenile retinoschisis. Ophthalmic Paediatr Genet 1985;5:13-7.
- 4. Sauer CG, Gehrig A, Warneke-Wittstock R, et al. Positional cloning of the gene associated with X-linked juvenile retinoschisis. Nat Genet 1997;17:164-70. PMID: 9326935.
- 5. Takada Y, Fariss RN, Muller M, et al. Retinoschisin expression and localization in rodent and human pineal and consequences of mouse RS1 gene knockout. Mol Vis 2006;12:1108-16.
- 6. Baumgartner S, Hofmann K, Chiquet-Ehrismann R, Bucher P. The discoidin family revisited: New members from prokaryotes and a homology based fold prediction. Protein Sci 1998;7:1626-31.
- 7. Vogel W. Discoidin domain receptors: structural relations and functional implications. FASEB J 1999;13:S77-82.
- 8. Functional implications of the spectrum of mutations found in 234 cases with X-linked juvenile retinoschisis. The Retinoschisis Consortium. Hum Mol Genet 1998;7:1185-92.
- 9. Grayson C, Reid SN, Ellis JA, et al. Retinoschisin, the X-linked retinoschisis protein, is a secreted photoreceptor protein, and is expressed and released by Weri-Rb1 cells. Hum Mol Genet 2000;9:1873-9.
- 10. Molday LL, Hicks D, Sauer CG, et al. Expression of X-linked retinoschisis protein RS1 in photoreceptor and bipolar cells. Invest Ophthalmol Vis Sci 2001;42:816-25.
- 11. Molday RS, Warren R, Kim TS. Purification and biochemical analysis of cGMP-gated channel and Na/Ca(2)-K exchanger of rod photoreceptors. Methods Enzymol 2000;315:831-47.
- 12. Molday RS, Kellner U, Bernhard HF, et al. X-linked juvenile retinoschisis: clinical diagnosis, genetic analysis, and molecular mechanisms. Prog Retin Eye Res 2012;31:195-212.
- 13. McKibbin M, Booth AP, George ND. Foveal ectopia in X-linked retinoschisis. Retina 2001;21(4):361-6.
- 14. George ND, Yates JR, Bradshaw K, Moore AT. Infantile presentation of X linked retinoschisis. Br J Ophthalmol 1995;79(7):653-7.
- 15. Mitamura Y, Miyanishi K, Shizukawa N, et al. A case of X-linked retinoschisis diagnosed in an infant. Retina 2003;23(5):731-2.
- 16. George ND, Yates JR, Moore AT. X-linked retinoschisis. Br J Ophthalmol 1995;79:697-702.
- 18. Apushkin MA, Fishman GA, Rajagopalan AS. Fundus findings and longitudinal study of visual acuity loss in patients with X-linked retinoschisis. Retina 2005;25(5):612-18.
- 19. Kellner U, Brummer S, Foerster MH, Wessing A. X-linked congenital retinoschisis. Graefes Arch Clin Exp Ophthalmol 1990;228(5):432-7.
- 20. George ND, Yates JR, Moore AT. Clinical features in affected males with X linkedretinoschisis.Arch Ophthalmol 1996;114(3):274-80.
- 21. Campbell JP, Skalet AH, Lauer AK. Vitreous veils associated with congenital X-linked retinoschisis. JAMA Ophthalmol 2015;133(8):151-5.
- 22. Ando A, Takahashi K, Sho K, et al. Histopathological findings of X-linked retinoschisis with neovascular glaucoma. Graefes Arch Clin Exp Ophthalmol 2000;238(1):1-7.
- 23. Greven CM, Moreno RJ, Tasman W. Unusual manifestations of X-linked retinoschisis. Discussion 226-218. Trans Am Ophthalmol Soc 1990;88:211-25.
- 24. Tasman W, Greven C, Moreno R. Nasal retinal dragging in X-linked retinoschisis. Graefes Arch Clin Exp Ophthalmol 1991;229(4):319-22.
- 25. Sorsby A, Klein M, Gann JH, Siggins G. Unusual retinal detachment possibly sex-linked. Br J Ophthalmol 1951;35(1):1-10.
- 26. Haider NB, Jacobson SG, Cideciyan AV, et al. Mutation of a nuclear receptor gene, NR2E3, causes enhanced S cone syndrome, a disorder of retinal cell fate. Nat Genet 2000;4:127-31.
- 27. Sohn EH, Chen FK, Rubin GS, et al. Macular function assessed by microperimetry in patients with enhanced S-cone syndrome. Ophthalmology 2010;117:1199-206.
- 28. Sharon D, Sandberg MA, Caruso RC, et al. Shared mutations in NR2E3 in enhanced S-cone syndrome, Goldmann-Favre syndrome, and many cases of clumped pigmentary retinal degeneration. Arch Ophthalmol 2003;121(9):1316-23.
- 29. Sikkink SK, Biswas S, Parry NR, et al. X-linked retinoschisis: an update. J Med Genet 2007;44(4):225-32.
- 30. Koh AH, Hogg CR, Holder GE. The incidence of negative ERG in clinical practice. Doc Ophthalmol 2001;102:19-30.
- 31. Renner AB, Kellner U, Cropp E, Foerster MH. Dysfunction of transmission in the inner retina: incidence and clinical causes of negative electroretinogram. Graefes Arch Clin Exp Ophthalmol 2006;244:1467-73.
- 32. Sobaci G, Erdem U, Uysal Y, et al. Negative electroretinogram in the differential diagnosis of malingering of night blindness in the military. Mil Med 2007;172:402-4.
- 33. Bastos AL, Freitas Bde P, Villas Boas O, Ramiro AC. Use of topical dorzolamide for patients with X-linked juvenile retinoschisis: case report. Arq Bras Oftalmol 2008;71(2):286-90.
- 34. Genead MA, Fishman GA, Walia S. Efficacy of sustained topical dorzolamide therapy for cystic macular lesions in patients with X-linked retinoschisis. Arch Ophthalmol 2010;128:190-7.
- 35. Ghajarnia M, Gorin MB. Acetazolamide in the treatment of X-linked retinoschisis maculopathy. Arch. Ophthalmol 2007;125:571-3.
- 36. Zhang L, Reyes R, Lee W, et al. Rapid resolution of retinoschisis with acetazolamide. Doc Ophthalmol 2015;131(1):63-70.
- 37. Ferrone PJ, Trese MT, Lewis H. Vitreoretinal surgery for complications of congenital retinoschisis. Am J Ophthalmol 1997;123:742-7.
- 38. Sobrin L, Berrocal AM, Murray TG. Retinal detachment 7 years after prophylactic schisis cavity excision in juvenile X-linked retinoschisis. Ophthalmic Surg Lasers Imaging 2003;34:401-2.
- 39. Zeng Y, Takada Y, Kjellstrom S, et al. RS-1 gene delivery to an adult RS1h knockout mouse model restores ERG b-wave with reversal of the electronegative waveform of X-linked retinoschisis. Invest Ophthalmol Vis Sci 2004;45:3279-85.
- 40. Min SH, Molday LL, Seeliger MW, et al. Prolonged recovery of retinal structure/function after gene therapy in an RS1h-deficient mouse model of X-linked juvenile retinoschisis. Mol Ther 2005;12:644-51.

 Dodaj do ulubionych
Dodaj do ulubionych
