


Co znajdziesz w artykule?
- Mukopolisacharydoza typu III (MPS III) – postępująca choroba neurodegeneracyjna
- Przebieg i obraz kliniczny choroby Sanfilippa
- Diagnostyka, postępowanie i rokowanie u pacjentów z MPS III
Spis treści
Lizosomalne choroby spichrzeniowe to grupa ponad 50 defektów metabolicznych uwarunkowanych genetycznie. Wszystkie są chorobami monogenowymi dziedziczonymi, z nielicznymi wyjątkami, w sposób autosomalny recesywny. U podłoża każdej z nich leży niedobór enzymu odpowiedzialnego za degradację produktów metabolizmu komórkowego, który skutkuje akumulacją nierozłożonego produktu w lizosomach i tym samym upośledza ich funkcję.
Przez wiele lat lizosomy uważane były za niezbyt istotną organellę komórkową
mającą za zadanie utylizację produktów przemian wewnątrzkomórkowych i odzyskiwanie cząsteczek budulcowych potrzebnych do dalszych przemian. Na przestrzeni ostatnich lat pogląd na rolę lizosomu w komórce bardzo się zmienił. W wielu badaniach dowiedziono, że ta organella stanowi centrum koordynujące wiele ścieżek sygnałowych w komórce, w tym jest kluczowym sensorem wewnątrzkomórkowych stężeń substancji odżywczych związanych ze szlakiem mTOR. W komórce odbywa się nieustanny obrót białek, a lizosomy zapewniają koordynację procesów degradacji i autofagii białek w zależności od potrzeb poszczególnych organelli 1 .
Defekt metaboliczny dotyczący enzymów lizosomalnych skutkuje zaburzeniami procesów na różnych poziomach. Spichrzanie nierozłożonego produktu jest najbardziej oczywistym, pierwszym etapem choroby. To, co dzieje się później, to kaskada zdarzeń prowadząca do upośledzenia funkcji całej komórki oraz jej otoczenia, gdyż wadliwie działająca komórka zaczyna produkować cytokiny prozapalne. Następnym etapem jest apoptoza komórki, jej rozkład w tkankach przez komórki fagocytarne, w których w dalszym ciągu dochodzi do akumulacji substratu i w związku z tym wydzielania cytokin prozapalnych w jeszcze większej ilości. Do tego następuje także wtórne zahamowanie rozkładu innych substratów w lizosomach, co dodatkowo wzmacnia proces zapalny.
Mukopolisacharydoza (MPS) typu III jest najczęściej rozpoznawanym typem mukopolisacharydozy, występuje z częstością mniej więcej 1 na 100 000 urodzeń. Jako pierwszy chorobę tę opisał w 1963 roku amerykański lekarz Sylwester Sanfilippo, od którego nazwiska nazywana jest chorobą Sanfilippa.
W przypadku MPS III dochodzi do zaburzenia degradacji siarczanu heparanu (HS – heparan sulfate) wewnątrz lizosomów. HS to polisacharyd o ujemnym ładunku (inaczej glikozoaminoglikan [GAG]), który przyłączany jest do ogromnej liczby białek znajdujących się zarówno na powierzchni, jak i w wewnątrzkomórkowym matriksie. HS w połączeniu z białkami tworzy proteoglikany, które katabolizowane są w lizosomach. W procesie tym bierze udział kilka enzymów, w związku z czym wyróżniamy 5 typów MPS III (A-E) zależnie od rodzaju uszkodzonego enzymu 2 (tab. 1).
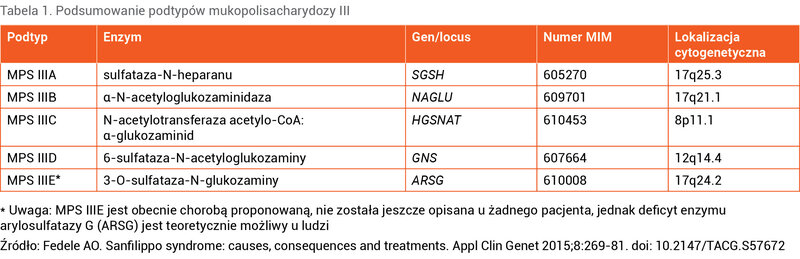
Tabela 1. Podsumowanie podtypów mukopolisacharydozy III
Akumulacja HS w ośrodkowym układzie nerwowym (OUN) powoduje lawinę wtórnych procesów patofizjologicznych, takich jak: reakcja zapalna i apoptoza neuronów, astocytoza, mikroglioza oraz dezorganizacja synaptyczna 3 .
Dokładny przebieg zdarzeń od momentu kumulacji HS do degeneracji OUN jest stosunkowo słabo poznany. Odkryto, że kumulacja HS hamuje wtórnie degradację gangliozydów GM2 i GM3, tym samym doprowadzając do wtórnego spichrzania tych związków zarówno w sposób bezpośredni, jak i poprzez negatywny wpływ HS na regulację syntezy gangliozydów. Wpływ akumulacji gangliozydów GM2 i GM3 na proces neurodegeneracji w przebiegu MPS III nadal jest przedmiotem badań, najbardziej prawdopodobny wydaje się jednak silny wpływ tych substancji na odpowiedź zapalną, która w znacznym stopniu przyspiesza degenerację neuronów OUN. Dodatkowo dowiedziono na modelach zwierzęcych, że w MPS III dochodzi w większym stopniu do spichrzania zarówno GAG, jak i gangliozydów w komórkach nerwowych niż w innych typach MPS 4 .
Przebieg kliniczny MPS III
Pogląd na przebieg kliniczny choroby Sanfilippa zmienił się w ostatnich latach. Dzięki powszechności diagnostyki genetycznej, zwłaszcza badań panelowych oraz całoegzomowego sekwencjonowania genomu (WES – whole-exome sequencing), coraz częściej rozpoznanie to jest ustalane u pacjentów z łagodniejszą postacią choroby, u których nie występują charakterystyczne cechy fenotypowe.
Tak jak każda lizosomalna choroba spichrzeniowa MPS III to spektrum fenotypów zależnych od aktywności resztkowej enzymu. Dla ułatwienia fenotypy te nazywane są: postacią ciężką, postacią łagodną lub postacią o późnym początku (attenuated form).
Najcięższa postać choroby jest związana z najmniejszą aktywnością resztkową enzymu (zwykle bliską zera). Jej przebieg można podzielić na 3 fazy poprzedzone okresem bezobjawowym trwającym pierwszych kilka lub kilkanaście miesięcy życia. W pierwszej fazie, która trwa od 1 do 3 roku życia dziecka, obserwuje się powolne zahamowanie rozwoju psychoruchowego (najbardziej wyraźne w sferze mowy, funkcje ruchowe są początkowo w pełni zachowane). Charakterystyczne objawy to: różnie nasilona dysmorfia (pogrubiałe rysy twarzy, synophrys, nadmierne owłosienie), powiększenie narządów wewnętrznych, sztywność stawowa. W pierwszych latach choroby dominują zaburzenia zachowania, które stopniowo ustępują wraz z postępem degeneracji OUN. W drugiej fazie choroby, która zaczyna się w 3-4 roku życia, obserwuje się: postępujące pogorszenie funkcji poznawczych, nasilenie zaburzeń zachowania i zaburzenia snu. Trzecia faza zaczyna się zwykle w wieku nastoletnim i objawia zaawansowanym otępieniem oraz utratą funkcji motorycznych. Problemy z zachowaniem stopniowo ustępują. Chorzy stają się całkowicie zależni od innych osób, tracą możliwość samodzielnego poruszania się, dołączają dysfagia i porażenie spastyczne, stan pacjentów określa się jako wegetatywny. Zgon następuje w drugiej dekadzie lub na początku trzeciej dekady życia 5 .
U pacjentów z pośrednią postacią choroby na pierwszy plan wysuwa się opóźnienie rozwoju z często towarzyszącymi zaburzeniami zachowania (takimi jak: nadpobudliwość, wybuchy złości, zachowania destrukcyjne, agresja). Opóźnienie rozwoju mowy zauważane jest około 4 roku życia. Zaburzenia zachowania podobnie jak zaburzenia snu występują niestale, zdarzają się okresy prawidłowego funkcjonowania. W tej grupie chorych w wywiadzie dominują nawracające infekcje dróg oddechowych we wczesnych latach życia. Hepatomegalia także nie jest charakterystyczna dla tej postaci choroby
W przypadku dorosłych osób powodem rozpoczęcia diagnostyki genetycznej były takie schorzenia, jak dystrofia siatkówki lub przerostowa kardiomiopatia, pacjenci ci nie prezentowali zaburzeń funkcji poznawczych 6 . Jest to przykład najłagodniejszego przebiegu klinicznego MPS III. W piśmiennictwie można znaleźć opisy przypadków pacjentów z demencją zdiagnozowanych w szóstej, a nawet w ósmej dekadzie życia, którzy prawidłowo funkcjonowali przez lata 7 .
Wydaje się, że wielu pacjentów z postacią o późnym początku nigdy nie zostało zdiagnozowanych, natomiast szczegółowe dane byłyby dostępne dopiero po wprowadzeniu przesiewowych badań noworodków w kierunku MPS III.
Trudności diagnostyczne
Mukopolisacharydozy jako choroby spichrzeniowe są zwykle kojarzone z konkretnymi cechami fenotypowymi, takimi jak: pogrubiałe rysy twarzy, niski wzrost, powiększona wątroba i śledziona, deformacje stawów. W MPS typu III objawy somatyczne są znacznie mniej nasilone, a w niektórych przypadkach mogą nie występować, co znacznie utrudnia postawienie właściwej diagnozy.
Spektrum objawów somatycznych u pacjentów z MPS III różni się nawet między rodzeństwem będącym nosicielami tych samych wariantów patologicznych w genie odpowiedzialnym za wystąpienie choroby (obserwacje własne). Bardzo często powodem zgłoszenia się rodziców do lekarza są zahamowanie rozwoju dziecka lub utrata umiejętności, które dziecko już opanowało. Do problemów z zachowaniem dzieci z MPS III należą: znaczna hiperaktywność, hiperoralność (skłonność do poznawania otoczenia za pomocą zmysłu smaku), upór, nieposłuszeństwo, częste napady złości, brak strachu przed zagrożeniem, a także zachowania destrukcyjne 8 . Objawy somatyczne u tych chorych mogą być bardzo subtelne, nawet przy nasilonych objawach ze strony OUN.
U pacjentów z łagodniejszą postacią MPS III objawy rozwijają się wolniej, a wśród nich dominują: zmniejszona męczliwość, nadwrażliwość na dotyk i zmiany temperatury, drażliwość, skłonność do płaczu, stereotypowa mowa, tendencja do agresywnego zachowania. Gdy pacjenci są w wieku nastoletnim, tego typu zachowania mogą zostać potraktowane jako typowe dla okresu dorastania, dopiero ich nasilenie w wieku dorosłym prowadzi do postawienia diagnozy 7 .
Kłopoty ze snem są niemal uniwersalne u dzieci z MPS III i wydają się korelować z nasileniem problemów z zachowaniem występujących w ciągu dnia. Podobnie jak u pacjentów z innymi schorzeniami neurozwyrodnieniowymi zaburzenia rytmu snu i czuwania są powiązane ze stopniem zaawansowania choroby. Wśród problemów ze snem występują zarówno trudności z zaśnięciem, budzenie się w nocy, okresowe pobudzenie w godzinach nocnych (krzyk, śpiewanie, śmiech), budzenie się bardzo wcześnie rano, jak i zasypianie w ciągu dnia. Uważa się, że problemy ze snem w tej grupie pacjentów wynikają ze zmienionego rytmu dobowego wydzielania melatoniny, polegającego na zmniejszeniu jej stężenia w nocy, a zwiększeniu w godzinach porannych w odniesieniu do grupy kontrolnej 8 . Stosowanie preparatów z melatoniną u tych pacjentów często przynosi poprawę w zakresie jakości snu.
W przypadku podejrzenia u dziecka choroby Sanfilippa należy oznaczyć wydalanie GAG w moczu oraz zbadać aktywność enzymów lizosomalnych w leukocytach krwi (w Polsce badanie to wykonywane jest w Pracowni Metabolicznej Instytutu Psychiatrii i Neurologii w Warszawie). W łagodnych postaciach choroby ustalenie rozpoznania jest możliwe jedynie na podstawie badań genetycznych, zatem w przypadku otrzymania prawidłowych wyników badań biochemicznych wskazana jest diagnostyka molekularna.
Opieka nad pacjentem z chorobą Sanfilippa
Ze względu na niezbyt nasilone objawy somatyczne dzieci z MPS III wykazują się prawidłową siłą fizyczną i sprawnością motoryczną, szczególnie w pierwszych latach po postawieniu diagnozy, co sprawia, że opieka nad nimi jest niezwykle trudna.
Rezonans magnetyczny (MR) mózgowia u chorych na MPS III uwidocznia atrofię kory mózgowej (od łagodnej do średnio nasilonej) oraz poszerzenie układu komorowego. Często opisywane są także zmiany płynowe o morfologii plastra miodu w istocie białej odpowiadające poszerzonym przestrzeniom okołonaczyniowym. Uważa się, że nasilenie zmian w obrazie MR mózgowia nie odpowiada ciężkości przebiegu klinicznego MPS III 9 , dlatego zasadność wykonywania tych badań kontrolnie w znieczuleniu ogólnym należy uznać za wątpliwą.
W swoim badaniu Barone i wsp. analizowali zapisy elektroencefalograficzne (EEG) u pacjentów z MPS III w korelacji z wiekiem i stopniem zaawansowania choroby. U dzieci poniżej 3 roku życia zapis EEG był prawidłowy, a po 6 roku życia obserwowano spowolnienie czynności podstawowej zwłaszcza w okolicy ciemieniowej, zmiany te były jeszcze bardziej wyraźne po ukończeniu 11 roku życia 10 .
Padaczka w przebiegu MPS III dotyczy do 45% chorych (MPS IIIA i IIIB). U zdecydowanej większości pacjentów występują napady uogólnione toniczno-kloniczne, z mniejszą częstością mogą pojawiać się także napady miokloniczne, nieświadomości. U pacjentów z chorobą Sanfilippa opisano także występowanie nocnej padaczki czołowo-skroniowej (NFLE – nocturnal frontal lobe epilepsy), z napadami hipermotorycznymi pod postacią gwałtownych obrotów tułowia, automatyzmów motorycznych i wokalizacji, występującymi wyłącznie we śnie. W badaniu EEG podczas napadu rejestrowane są zespoły fala ostra–fala wolna 4-6 Hz w odprowadzeniach czołowych, epizody trwają 5-15 s i powtarzają się wielokrotnie w ciągu nocy 11 . Tego typu padaczka w znacznym stopniu zakłóca sen i w konsekwencji jeszcze bardziej zaburza funkcjonowanie chorego dziecka.
Bardzo często ustalenie rozpoznania padaczki sprawia dużo trudności ze względu na całościowe zaburzenia rozwojowe typowe dla obrazu choroby. Występowanie napadów nieświadomości lub niedrgawkowych stanów padaczkowych może być niewłaściwie zinterpretowane przez opiekunów, podobnie jak ogniskowy początek napadu padaczkowego.
Leczenie padaczki u dzieci z MPS III nie różni się od leczenia innych pacjentów z padaczką. Po postawieniu diagnozy padaczki optymalne jest stosowanie monoterapii, po dodaniu kolejnego leku przewidywana poprawa kontroli napadów występuje jedynie u 10% pacjentów. Dobrą kontrolę napadów po włączeniu leczenia przeciwpadaczkowego uzyskuje się u 86% pacjentów z MPS III i padaczką. W tej grupie u 70% leczonych dzieci odnotowano brak napadów, a u 30% uzyskano redukcję częstości ich występowania 12 .
Podczas gdy napady uogólnione toniczno-kloniczne w znacznej większości całkowicie poddają się leczeniu, to w przypadku napadów ogniskowych nie obserwuje się takiej skuteczności leczenia 13 .
Należy mieć na uwadze, że chociaż zachowanie dzieci chorych na MPS III wynika głównie z procesów neurodegeneracyjnych, to należy pamiętać o innych przyczynach związanych z chorobą podstawową, które także mogą wpływać na zachowanie dziecka i zaburzenia snu. Przykładowo powiększenie narządów wewnętrznych może leżeć u podłoża niepokoju i mieć bezpośredni wpływ na zachowanie dziecka. Niedosłuch wynikający z przerostu tkanki limfatycznej gardła może wpływać negatywnie na rozwój mowy. Z powodu problemów z zachowaniem dziecka opiekunowie mogą nie zwrócić uwagi na napady padaczkowe, które nieleczone w oczywisty sposób pogarszają funkcjonowanie chorego 11 .
Optymalnie w procesie diagnostycznym padaczki u dzieci z MPS III wykonuje się badanie EEG oraz polisomnografię (PSG) w celu wykluczenia nocnego bezdechu sennego jako przyczyny zaburzeń snu. Zaleca się przeprowadzenie obu badań u pacjenta po ustaleniu rozpoznania choroby, a następnie powtarzanie ich raz w roku 14 . W praktyce jest to bardzo trudne z powodu często bardzo nasilonych zaburzeń zachowania i braku współpracy pacjentów.
Obturacyjny bezdech senny pogłębia istniejące już problemy ze snem, a następnie funkcjonowanie dziecka w ciągu dnia 15 . Pacjenci wymagają stałej opieki laryngologicznej, zdarzają się nawroty po wykonanych adenotomii i tonsillektomii wymagające ponownego leczenia operacyjnego.
Do zajęcia serca u pacjentów z chorobą Sanfilippa dochodzi rzadziej niż w przebiegu innych typów MPS i zwykle powikłania kardiologiczne są mniej nasilone. Niemniej u 40% chorych może rozwinąć się dysfunkcja zastawek, najczęściej niedomykalność mitralna lub aortalna, zazwyczaj łagodna 16 . Wiąże się z tym konieczność kontrolnego wykonywania badania echokardiograficznego w tej grupie pacjentów optymalnie raz w roku lub według bieżących zaleceń kardiologicznych.
W piśmiennictwie dostępne są opisy przypadków dorosłych pacjentów z MPS III, u których wystąpił całkowity blok przedsionkowo-komorowy, w konsekwencji czego nastąpił nagły zgon chorego 17 .
Osoby z chorobą Sanfilippa mogą także wymagać opieki ortopedycznej. Zmiany kostno-stawowe wynikające z procesu spichrzania GAG wymagające interwencji chirurgicznej to najczęściej: zespół cieśni nadgarstka, przykurcze palców oraz postępująca skolioza dużego stopnia. Stosunkowo często w tej grupie pacjentów występuje jałowa martwica głowy kości udowej mogąca powodować znaczne dolegliwości bólowe, które w sytuacji utrudnionego kontaktu z pacjentem i postępującego charakteru choroby, w której obraz wpisana jest stopniowa utrata mobilności, mogą pozostać nierozpoznane przez dłuższy czas 18 .
Podsumowanie
W przeciwieństwie do innych typów mukopolisacharydoz, w przypadku których pacjenci wykazują znacznie nasilone objawy somatyczne, u chorych na MPS III występują głównie zaburzenia funkcji poznawczych oraz objawy neurologiczne. Niezależnie od postaci MPS III opóźnienie rozwoju mowy jest obecne praktycznie w każdym przypadku 19 . Objawy somatyczne, takie jak: pogrubiałe rysy twarzy, wydatne brwi, sztywne włosy, hepatomegalia, zmiany stawowe, mogą być bardzo subtelne, ale w przypadku ich stwierdzenia należy skierować pacjenta na dalszą diagnostykę.
Prawdziwym wyzwaniem są osoby z łagodniejszą postacią choroby, u których nie występują cechy dysmorfii. Izolowany opóźniony rozwój mowy z prawidłowym rozwojem w innych sferach nie jest sytuacją rzadką, dlatego u chorych na MPS III często rozpoznaje się idiopatyczne opóźnienie rozwoju mowy. W przypadku innych pacjentów, u których występują cechy całościowych zaburzeń rozwojowych, często stawiana jest diagnoza zaburzeń ze spektrum autyzmu. W przypadku pacjentów prezentujących znaczną nadruchliwość, pobudzenie, impulsywność nietrudno o ustalenie rozpoznania zespołu nadpobudliwości psychoruchowej z deficytem uwagi (ADHD – attention-deficit hyperactivity disorder).
To, co wyróżnia dzieci z chorobą Sanfilippa, to przede wszystkim bardzo powolny postęp rozwoju lub jego regres oraz problemy ze snem.
Obecnie nie ma dostępnych terapii enzymatycznych dla pacjentów z MPS III. Ze względu na to, że proces chorobowy toczy się głównie w OUN, ograniczona penetracja substancji terapeutycznych przez barierę krew–mózg jest ogromnym problemem. Prowadzonych jest wiele badań klinicznych dotyczących leczenia choroby Sanfilippa, a największe nadzieje wiązane są z terapiami genowymi. Wstępne wyniki tych badań są bardzo obiecujące, kluczowe jest jednak jak najwcześniejsze wdrożenie leczenia. Dlatego potrzeba skutecznej i niezwłocznej diagnostyki w przypadku podejrzenia choroby Sanfilippa.
Abstract
The diagnostic odyssey of patients with Sanfilippo disease
Mucopolysaccharidosis type III (Sanfilippo Disease) is the most common mucopolysaccharidosis. Diagnostic problems are related to the presence of benign somatic symptoms. The disease mainly involves the central nervous system, so the major symptoms comprise speech delay, hyperactivity and sleep disorders. Patients can also develop epilepsy and cardiologic, orthopedic and otolaryngologic problems. Patients need an interdisciplinary approach. Because of the progressive nature of the disease, diagnosis should be made as early as possible.
- 1. Lamming DW, Bar-Peled L. Lysosome: the metabolic signaling hub. Traffic 2019;20(1):27-38. doi: 10.1111/tra.12617
- 2. Fedele AO. Sanfilippo syndrome: causes, consequences and treatments. Appl Clin Genet 2015;8:269-81. doi: 10.2147/TACG.S57672
- 3. Bigger BW, Begley DJ, Virgintino D, et al. Anatomical changes and pathophysiology of the brain in mucopolysaccharidosis disorders. Mol Genet Metab 2018;125(4):322-31. doi: 10.1016/j.ymgme.2018.08.003
- 4. McGlynn R, Dobrenis K, Walkley SU. Differential subcellular localization of cholesterol, gangliosides and glycosaminoglycans in murine models of mucopolysaccharide storage disorders. J Comp Neurol 2004;480(4):415-26. doi: 10.1002/cne.20355
- 5. Meyer A, Kossow K, Gal A, et al. Scoring evaluation of the natural course of mucopolysaccharidosis type IIIA (Sanfilippo syndrome type A). Pediatrics 2007;120(5):e1255-61. doi: 10.1542/peds.2007-0282
- 6. Nijmeijer SCM, van den Born LI, Kievit AJA, et al. The attenuated end of the phenotypic spectrum in MPS III: from late-onset stable cognitive impairment to a non-neuronopathic phenotype. Orphanet J Rare Dis 2019;14(1):249. doi: 10.1186/s13023-019-1232-0
- 7. Moog U, van Mierlo I, van Schrojenstein Lantman-de Valk HM, et al. Is Sanfilippo type B in your mind when you see adults with mental retardation and behavioral problems? Am J Med Genet C Semin Med Genet 2007;145C(3):293-301. doi: 10.1002/ajmg.c.30142
- 8. Mahon LV, Lomax M, Grant S, et al. Assessment of sleep in children with mucopolysaccharidosis type III. PLoS One 2014;9(2):e84128. doi: 10.1371/journal.pone.0084128
- 9. Palmucci S, Attinà G, Lanza ML, et al. Imaging findings of mucopolysaccharidoses: a pictorial review. Insights Imaging 2013;4(4):443-59. doi: 10.1007/s13244-013-0246-8
- 10. Barone R, Cocuzza MD, Guida C, et al. EEG features in patients with mucopolysaccharidoses III at different disease stages. J Inherit Metab Dis 2016;39(Suppl 1):S186
- 11. Bonanni P, Volzone A, Randazzo G, et al. Nocturnal frontal lobe epilepsy in mucopolysaccharidosis. Brain Dev 2014;36(9):826-9. doi: 10.1016/j.braindev.2013.12.002
- 12. Delgadillo V, O'Callaghan Mdel M, Gort L, et al. Natural history of Sanfilippo syndrome in Spain. Orphanet J Rare Dis 2013;8:189. doi: 10.1186/1750-1172-8-189
- 13. Barone R, Pellico A, Pittalà A, et al. Neurobehavioral phenotypes of neuronopathic mucopolysaccharidoses. Ital J Pediatr 2018;44(Suppl 2):121. doi: 10.1186/s13052-018-0561-2
- 14. Mariotti P, Della Marca G, Iuvone L, et al. Sleep disorders in Sanfilippo syndrome: a polygraphic study. Clin Electroencephalogr 2003;34(1):18-22. doi: 10.1177/155005940303400108. PMID: 12515448.
- 15. Leighton SE, Papsin B, Vellodi A, et al. Disordered breathing during sleep in patients with mucopolysaccharidoses. Int J Pediatr Otorhinolaryngol 2001;58(2):127-38. doi: 10.1016/ s0165-5876(01)00417-7
- 16. Lin HY, Chen MR, Lin SM, et al. Cardiac characteristics and natural progression in Taiwanese patients with mucopolysaccharidosis III. Orphanet J Rare Dis 2019;14(1):140. doi: 10.1186/s13023-019-1112-7
- 17. Misumi I, Chikazawa S, Ishitsu T, et al. Atrioventricular block and diastolic dysfunction in a patient with Sanfilippo C. Intern Med 2010;49(21):2313-6. doi: 10.2169/internalmedicine.49.4210
- 18. White KK, Karol LA, White DR, et al. Musculoskeletal manifestations of Sanfilippo Syndrome (mucopolysaccharidosis type III). J Pediatr Orthop 2011;31(5):594-8. doi: 10.1097/BPO.0b013e31821f5ee9
- 19. Héron B, Mikaeloff Y, Froissart R, et al. Incidence and natural history of mucopolysaccharidosis type III in France and comparison with United Kingdom and Greece. Am J Med Genet A 2011;155A(1):58-68. doi: 10.1002/ajmg.a.33779
Następny artykuł:

 Dodaj do ulubionych
Dodaj do ulubionych
