



Spis treści
Celem poniższej pracy jest przedstawienie aktualnych osiągnięć w dziedzinie genetyki OBL u dzieci i dorosłych w świetle ich klinicznego znaczenia oraz roli tych badań na tle innych metod diagnostycznych stosowanych w rutynowej praktyce laboratoryjnej
WPROWADZENIE
Ostre białaczki limfoblastyczne (OBL) są heterogenną grupą chorób nowotworowych dotyczącą niedojrzałych komórek limfoidalnych, które naciekają szpik kostny oraz krew, a także mogą tworzyć nacieki w różnych organach. OBL z różną
częstością występują u osób w każdej grupie wiekowej, ale u dzieci stanowią 80 proc. wszystkich białaczek, ze szczytem zachorowania pomiędzy 2. a 5. r.ż. U dorosłych OBL stanowią 20 proc. wszystkich ostrych białaczek, zaś ich występowanie obserwuje się przeważnie przed 30. r.ż.. 1 W OBL u dzieci uzyskuje się wyleczenie u 3/4 chorych. Natomiast pomimo dużego postępu w dziedzinie leczenia OBL, u większości dorosłych i niemowląt terapia kończy się niepowodzeniem, co oznacza śmierć pacjenta. 2
Ten fakt wzmacnia konieczność poznania mechanizmów transformacji OBL, a następnie modyfikacji aktualnego systemu klasyfikacyjnego, tak aby umożliwić podejmowanie prób opracowania nowych form terapii, lepiej dostosowanych do choroby u danej osoby. Terapie te, poprzez ingerencję w mechanizmy prowadzące do rozwoju choroby, byłyby bardziej skuteczne w zabijaniu komórek białaczkowych, a przy tym mniej toksyczne dla komórek zdrowych. Aby ustalić wskazania do takich terapii, niezbędny byłby indywidualny dobór pacjentów, możliwy do przeprowadzenia na podstawie metod diagnostycznych, które obecnie stosowane są wyłącznie w celach badawczych.
DOTYCHCZASOWE SYSTEMY KLASYFIKACYJNE W OBL
Podstawowy podział OBL dokonywany jest na podstawie dominującego w obrazie rodzaju komórki, stąd OBL zostały następnie podzielone na:
- białaczkę B-prekursorową (B-OBL),
- T-prekursorową (T-OBL). 1
Pierwotny podział B-OBL w ramach klasyfikacji FAB (ang. French-American-British) ma już tylko znaczenie historyczne. Podział EGIL dla T-OBL (ang. European Group for the Immunological Classification of Leukemias), opierający się na wyniku badania immunofenotypowego, określa fizjologiczny etap różnicowania limfocytu, któremu odpowiada dominujący liczbowo rodzaj komórek białaczkowych. Liczne badania potwierdzają jednak, że u podłoża poszczególnych podtypów białaczek, które zostały zdefiniowane na podstawie cytomorfologii oraz immunofenotypu, leżą swoiste zaburzenia genetyczne.
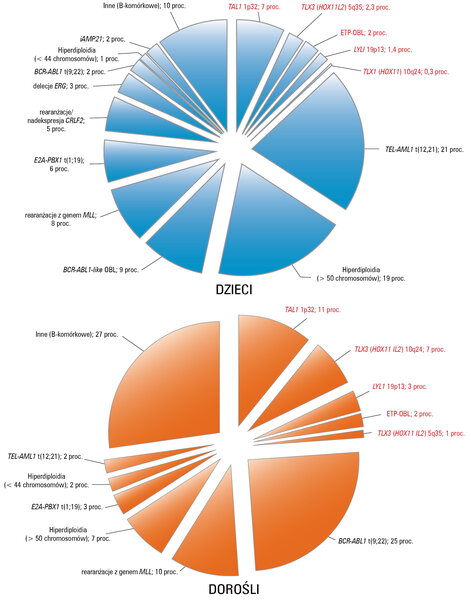
Ryc. 1. Wykresy przedstawiają częstotliwość występowania poszczególnych zmian cytogenetycznych i genetycznych w B-OBL (czarny tekst) i T-OBL (czerwony tekst) u dzieci i dorosłych. Wyjaśnienia skrótów w tekście.
W tej sytuacji kolejne systemy klasyfikacyjne, w tym aktualna wersja WHO 2008, 3 uwzględniają wybrane zmiany genetyczne jako podstawowe kryterium definiowania podjednostek choroby. Dzięki uwzględnieniu zaburzeń cytogenetycznych w kryteriach podziału, klasyfikacja WHO 2008 pozwala w sposób bardziej precyzyjny, niż czyniły to poprzednie wersje FAB i EGIL ustalić ryzyko nawrotu choroby.
Ponieważ klasyfikacja ta bierze pod uwagę jedynie duże zmiany chromosomalne, w założeniu nie jest doskonała, gdyż nie umożliwia subklasyfikacji dużej grupy pacjentów z kariotypem prawidłowym. Dalsza diagnostyka oraz różnicowanie ryzyka nawrotu choroby tej grupy stają się możliwe dzięki zastosowaniu bardziej zaawansowanych metod badań genetycznych, które poddają analizie cały genom: wysokorozdzielcze techniki, oparte głównie na technice mikromacierzy i łańcuchowej reakcji polimerazy (PCR). Za pomocą tych metod, w grupie pacjentów bez dużych aberracji chromosomalnych – w ostatnich kilku latach wykryto obecność wielu zmian na poziomie pojedynczych nukleotydów (tzw. rearanżacje wewnątrzgenowe). 4 Dodatkowo okazuje się również, że zmiany te towarzyszą wielu dużym translokacjom chromosomalnym i ich obecność różnicuje rokowanie w danym cytogenetycznym podtypie OBL. Ostatnie badania wyjaśniają również mechanizmy niestabilności genetycznej w OBL, która prowadzi do rozwoju wtórnych aberracji genetycznych i przyczynia się do fatalnego przebiegu wielu podtypów białaczek.
Dzięki badaniom obejmującym analizę całego genomu, lista nowych aberracji genetycznych w OBL z roku na rok się wydłuża. Stworzenie zatem idealnej klasyfikacji, która uwzględniałaby wszystkie zaburzenia genetyczne i jednocześnie korelowała z klinicznym przebiegiem choroby, wydaje się być niezmiernie trudne. Niezbędne zatem jest całościowe podejście do zagadnienia transformacji OBL, uwzględniające pojawienie się tzw. aberracji pierwotnych, mających kluczowe znaczenie dla rozwoju patologii (ang. driver mutations), jak również dynamikę procesu nabywania mutacji wtórnych, w tym pojawianie się mutacji mniej istotnych dla procesu transformacji (ang. passenger mutations).
GENETYCZNA CHARAKTERYSTYKA B-OBL
Na podstawie klasyfikacji WHO w OBL wywodzącej się z linii B-komórkowej wyróżnia się podklasy cytogenetyczne związane z występowaniem dużych aberracji chromosomalnych: hiperdiploidii (> 50 chromosomów), hipodiploidii (< 50 chromosomów) oraz translokacji: t(9;22)(q34;q11.2); BCR-ABL1, t(12;21);(p13;q22); TEL-AML1, t(1;19); (q23;p13.3); E2A-PBX1, t(5;14)(q31;q32); IL3-IGH, a także rearanżacji genu MLL t(v;11q23). 3
B-OBL z genem fuzyjnym BCR-ABL1
W wyniku wymiany części ramion długich pomiędzy chromosomem 9 a chromosomem 22 powstaje tzw. chromosom Filadelfia, zawierający gen fuzyjny BCR-ABL1 kodujący białko o aktywności kinazy tyrozynowej. Nadekspresja tego białka jest charakterystyczna dla chorych z przewlekłą białaczką szpikową (PBSz), a także 25 proc. OBL u dorosłych i 2-5 proc. OBL u dzieci. 5
Klinicznie pacjenci z OBL BCR-ABL1-dodatni są starsi, mają większą liczbę blastów we krwi obwodowej i zaliczani są do grupy wysokiego ryzyka: otrzymują intensywniejszą chemioterapię oraz w miarę możliwości są kwalifikowani do procedur przeszczepienia komórek macierzystych krwi w pierwszej remisji. 6
Wyniki leczenia istotnie zmieniło wprowadzenie inhibitorów kinaz tyrozynowych (imatynib, dazatynib, nilotynib), dzięki którym rokowanie chorych bardzo się poprawiło. Okazuje się jednak, że grupa pacjentów BCR-ABL1-dodatnich nie jest jednorodna, jeśli chodzi o dynamikę postępu choroby. Badania Cazzaniga i wsp. na bliźniętach jednojajowych, które posiadały taką samą chorobę OBL BCR-ABL1-dodatnią wykazały, że pomimo wspólnej cechy, jaką jest obecność białka BCR-ABL1, kliniczny przebieg choroby może diametralnie różnić się w poszczególnych przypadkach. W badaniu tym jedno z dwójki bliźniąt w każdej z par zmarło wskutek szybkiego nawrotu choroby, mimo zastosowania transplantacji komórek macierzystych, podczas gdy drugie z bliźniąt nie przejawiało tak agresywnego przebiegu choroby. Czynnikiem różnicującym i wpływającym na ewolucję białaczki okazała się obecność mutacji punktowych/delecji genu IKZF1: stwierdzono bowiem, że bliźnięta, które zmarły z powodu nawrotu choroby, posiadały takie mutacje, podczas gdy te, które przeżyły – nie. 7
Gen IKZF1 koduje bardzo ważny czynnik transkrypcyjny – IKAROS, biorący udział w rozwoju limfocytów B. Delecje IKZF1 często pojawiają się u pacjentów B-OBL BCR-ABL1-dodatnich (ponad 80 proc.). 8 Ostatnie badania wykazują, że są one obecne także u 1/3 pacjentów pediatrycznych B-OBL-negatywnych pod kątem genu BCR-ABL, u których nie stwierdza się także obecności innych genów fuzyjnych (kariotyp prawidłowy). 9 W każdym z przypadków ich obecność związana jest z krótszym całkowitym przeżyciem oraz większym ryzykiem nawrotu choroby.
Podgrupa BCR-ABL1-like OBL
Badania den Boer i wsp. 9 wykazały, że profil ekspresji genów w wybranej podgrupie osób, u których nie stwierdzono obecności genu fuzyjnego BCR-ABL1, ale stwierdzono obecność mutacji w genie IKZF, wykazuje podobieństwo do profilu ekspresji genów u pacjentów BCR-ABL1-dodatnich. Podgrupa tych pacjentów została zatem nazwana BCR-ABL1-podobna (ang. BCR-ABL1-like) i tak jak w przypadku pacjentów BCR-ABL1-dodatnich, jest ona związana z gorszym rokowaniem.
Dalsze badania wykazały, że wraz z mutacjami w genie IKZF1 współwystępuje również nadekspresja genu CRLF2 (ang. cytokine-receptor-like factor), do której dochodzi w wyniku rearanżacji tego genu, oraz mutacje genów JAK1 i JAK2 (ang. Janus kinase 1 i 2). 10, 11 Zgodnie z ostatnimi doniesieniami, że w grupie tych pacjentów obserwuje się również mutacje aktywujące geny innych kinaz tyrozynowych, np. ABL1 lub receptorów dla cytokin takich jak: EPOR, IL7R, PDGFRB. 12
Białaczka BCR-ABL1-podobna stanowi ok. 20 proc. dziecięcych OBL, czyli u pacjentów pediatrycznych jest 10 razy częstsza niż BCR-ABL1-dodatnia OBL. 13
Ponieważ zaburzone mechanizmy molekularne w tych białaczkach prowadzą do aktywacji ścieżki JAK-STAT, może to stanowić w przyszłości ważny punkt uchwytu dla terapii celowanych. Dotychczas wykonane próby przedkliniczne wykazały, że komórki z rearanżacjami genów ABL1 i PDGFRB są wrażliwe na inhibitory kinaz tyrozynowych, takie jak imatynib czy dazatynib. Natomiast inhibitory kinaz JAK mogą być skuteczne w przypadku rearanżacji genów JAK2 i IL7R. 12 Ciekawa jest obserwacja, że obydwa podtypy białaczek BCR-ABL1-dodatnich, jak również BCR-ABL1-podobnych, charakteryzują się szybszym tempem ewolucji klonalnej, co potwierdza obecność wtórnych mutacji towarzyszących. Wskazuje to na działanie czynników wpływających na niestabilność genetyczną komórek białaczkowych. 14
Ostatnie badania sekwencji punktów cięcia delecji w genie IKZF1 (ang. breakpoint regions) wykazały w ich pobliżu obecność sekwencji genomowych wskazujących na zaangażowanie genów kodujących białka aktywujące rekombinację tzw. RAG1/2 (ang. recombination activing gene1/2) 4 – enzymu wprowadzającego podwójne pęknięcia w DNA podczas rearanżacji łańcuchów immunoglobulin (Ig). Fakt ten sugeruje zaangażowanie fizjologicznych procesów w komórce limfocytu w progresję białaczki limfoblastycznej B-komórkowej. 15
B-OBL z rearanżacjami MLL
Gen MLL (ang. Mixed Lineage Leukemia) w stwierdzonych translokacjach chromosomowych może być partnerem dla ponad 80 różnych genów partnerskich. Białko to bierze udział w hematopoezie poprzez utrzymywanie prawidłowej ekspresji genów homeotycznych (HOX). 16 Rearanżacje z udziałem genu MLL są podłożem bardzo agresywnej postaci OBL CD10– u noworodków i stanowi ona 80 proc. przypadków w tej grupie wiekowej. 17 Natomiast w grupie dorosłych z OBL ich odsetek jest znacznie niższy, często związany z wcześniej stosowaną chemioterapią inhibitorami topoizomerazy II. 16, 18
Pięć najczęściej występujących translokacji z udziałem genu MLL (nazwa powstałego genu fuzyjnego jest podana po nazwie translokacji) to: t(4;11); MLL-AF4, t(9;11); MLL-AF9, t(11;19); MLL-ENL, t(10;11); MLL-AF10 oraz t(6;11); MLL-AF6. 19
Niektóre z nich mogą występować nie tylko w OBL, ale także w rozrostach mieloidalnych, np. w ostrej białaczce szpikowej (OBSz). Nieobecność bądź niewielka liczba dodatkowych mutacji świadczą o tym, że geny fuzyjne z udziałem genu MLL mają mocny potencjał transformujący komórki krwiotwórcze i nie wymagają wtórnych rearanżacji. Białaczki te wykazują również odrębny profil genetyczny i epigenetyczny. 20
B-OBl Z genem fuzyjnym TEL-AML1
Translokacja t(12;21);(p13;q22) prowadząca do powstania genu TEL-AML1 jest najczęściej występującą zmianą genetyczną w dziecięcej OBL, osiągając szczyt występowania u chorych pomiędzy 1. i 10. r.ż. 21, 22 Gen fuzyjny TEL-AML1 z dużą częstotliwością jest wykrywany u noworodków, lata przed ujawnieniem się pełnoobjawowej OBL, co sugeruje z jednej strony, że do jego powstania dochodzi już w wieku prenatalnym, a także, że do pełnej transformacji nowotworowej wymagane są dodatkowe zmiany genetyczne. 23
Badania potwierdziły, że genowi fuzyjnemu TEL-AML1 towarzyszą submikroskopowe aberracje, włączając w to delecje genów PAX5 i EBF1, jak również delecje drugiej kopii genu TEL. Okazało się, że mutacje wtórne, które występują znacznie częściej w TEL-AML1-pozytywnych OBL niż w innych podtypach białaczek, powstają w toku progresji choroby, jako wyraz niestabilności genetycznej i ewolucji klonalnej, która jej towarzyszy. Dlatego obecność genu TEL-AML1, chociaż wiąże się ze stosunkowo dobrym rokowaniem, może prowadzić do odległych nawrotów choroby u dzieci z jej remisją. 24 Komórki powodujące te nawroty, oprócz obecności tego samego genu fuzyjnego TEL-AML1, często cechują się zmienionym fenotypem w porównaniu z wyjściowym klonem białaczkowym (nowe łącze VDJ w receptorach IgH/TCR, a także immunofenotypowe markery powierzchniowe). Analiza sekwencji genomowych punktów cięcia w obrębie aberracji wtórnych sugeruje, że do ich wystąpienia doszło, podobnie jak w przypadku mutacji towarzyszących BCR-ABL1 – w wyniku aktywności białek RAG1/2.
Wewnątrzchromosomowa amplifikacja chromosomu 21
Wewnątrzchromosomowa amplifikacja chromosomu 21 (iAMP21) jest rzadką zmianą na poziomie cytogenetycznym występującą u około 2 proc. dzieci z B-OBL i jest definiowana jako co najmniej trzykrotne zwielokrotnienie regionu chromosomu 21 zawierającego gen AML1. 25 Amplifikacja iAMP21 pojawia się częściej u starszych dzieci i młodych dorosłych i jest związana ze złym rokowaniem. 26
B-OBL z delecją genu ERG
Najnowsze badania wykazują, że mikrodelecje w genie ERG (ang. ETS-related gene) są związane z dobrym rokowaniem u dzieci i występują u chorych z kariotypem prawidłowym. Cechą charakterystyczną tej podgrupy B-OBL jest:
- podobna morfologia blastów w badaniu mikroskopowym,
- ekspresja markera CD2,
- częste inne submikroskopowe aberracje, jak delecje w genie IKZF1. 27
Ostatnie badania wykazały, że pacjentów z B-OBL z mikrodelecją w genie ERG charakteryzuje unikatowy profil ekspresji genów. 28
GENETYCZNA CHARAKTERYSTYKA T-OBL
Białaczki te stanowią mniejszość zarówno wśród OBL dzieci, jak i dorosłych. Jednocześnie fenotyp T-komórkowy OBL jest związany z gorszym rokowaniem niż B-komórkowy. W porównaniu również z innymi rozrostami wywodzącymi się z komórek krwiotwórczych, proces transformacji prowadzący do T-OBL wydaje się być bardziej złożony i wieloetapowy.
Aberracje genetyczne związane są przynajmniej z czterema różnymi procesami komórkowymi:
- różnicowania,
- regulacji cyklu komórkowego,
- proliferacji,
- zdolności do samoodnowy (ang. self-renewal capacity).
Aberracje te współwystępują równolegle w tej samej komórce, ich produkty współpracują ze sobą i prowadzą do fatalnego w skutkach efektu transformacji i progresji choroby.
W przeciwieństwie do B-OBL, w T-OBL dużo częściej obserwujemy rearanżacje związane z locus kodującym swoisty receptor tych limfocytów, tj. T – TCR (ponad 30 proc. T-OBL). Ich efektem może być nieprawidłowa ekspresja onkogenów biorących udział w dojrzewaniu limfocytu T: TLX1 (HOX11), TLX3 (HOX11L2), LYL1, TAL1. 29 Fakt, że genem partnerskim w stosunku do wymienionych onkogenów jest TCR świadczy o tym, że geneza tych aberracji genetycznych związana jest ściśle z etapem dojrzewania limfocytów T, w którym dochodzi do rearanżacji receptorów TCR (β-selekcja).
Obok tych rearanżacji, w T-OBL występują także geny fuzyjne niezwiązane z receptorem TCR (np. SIL-TAL, CALM-AF10, NUP214-ABL) oraz mutacje pojedynczych nukleotydów.
Wieloetapowość procesu transformacji T-OBL podkreśla m.in. fakt częstego współistnienia mutacji np. w genie NOTCH1 lub delecji p16/INK4A oraz p14/ARF z innymi rearanżacjami genetycznymi u ok. odpowiednio 50 i 70 proc. chorych T-OBL – bez wyraźnej preferencji, jeśli chodzi o rodzaj towarzyszącej aberracji.
Wyniki badań wskazują, że podgrupy białaczek T-OBL według typu aberracji genetycznej nie są tak mocno związane z rokowaniem, jak w przypadku B-OBL. Związek z rokowaniem natomiast wykazują podgrupy T-OBL utworzone w ramach klasyfikacji, która uwzględnia fizjologiczne etapy dojrzewania limfocytu T 30, 31, 32 : EGIL oraz podział wg Asnafi i wsp. 33 Szczególnie ten ostatni w oparciu o wynik badania klonalności immunofenotypu TCR dzieli T-OBL na formę: niedojrzałą, pre-α/β, oraz dojrzałą α-β i γ-δ. Pośród wymienionych białaczek, OBL pre-α/β odpowiada etapowi fizjologicznej β-selekcji i przejawia dobre rokowanie.
T-OBL z genem fuzyjnym SIL-TAL1
Translokacja SIL-TAL1 jest jedną z najczęstszych translokacji w T-OBL, szczególnie u dzieci. Jej efektem jest nadekspresja genu TAL1. 34 Białaczki te cechują się odrębną charakterystyką:
- dużą liczbą białych krwinek (WBC),
- stężeniem hemoglobiny (Hb),
- ekspresją markerów CD2.
Rola tej translokacji w aspekcie rokowniczym zostaje nadal niejasna. 35, 36, 37
T-OBL z genem fuzyjnym CALM-AF10
Translokacja t(10;11) występuje dość rzadko w T-OBL. Prace Asnafi i wsp. 38 wskazują, że gen fuzyjny CALM-AF10 występuje w dwóch izoformach: izoforma CALM-AF10, związana z niedojrzałym fenotypem limfocytów T, cechuje się niekorzystnym rokowaniem, natomiast T-OBL CALM-AF10-dodatnia, wykazująca fenotyp TCR γ/δ, ulegała stosunkowo szybkiej całkowitej remisji. 38
T-OBL z genem fuzyjnym NUP214-ABL
NUP214-ABL1 jest obserwowany u ok. 6 proc. pacjentów z T-OBL, i co ciekawe, ostatnio został również odnotowany w podgrupie B-OBL BCR-ABL-like. 12 Podobnie jak w przypadku genu fuzyjnego BCR-ABL1, gen NUP214-ABL1 jest odpowiedzialny za konstytutywną aktywację kinazy tyrozynowej ABL1, co sprawia, że jest wrażliwy na inhibitory kinaz tyrozynowych. 39
Mutacje aktywujące gen NOTCH1
Konsekwencją mutacji aktywujących NOTCH1 w T-OBL jest nadmierna ekspresja tego białka. 40 Ponadto u ok. 15 proc. pacjentów występują dodatkowe mutacje genu FBXW7, które wzmagają aktywność białka NOTCH1. 41 Obecność tych mutacji stała się punktem wyjścia do prac nad terapią celowaną. 42 Badania kliniczne wykazały lepsze ogólne przeżycie chorych z mutacją NOTCH1 (OS – ang. overall survival). 43
T-OBL z wczesnych komórek prekursorowych limfocytów T (ETP-OBL)
Niedawno została opisana grupa pacjentów T-OBL o bardzo niekorzystnym rokowniczo profilu genetycznym. 44
ETP-OBL cechuje się również dużą liczbą zmian genetycznych, w tym tych charakterystycznych dla linii mieloidalnej, a więc przypominających ostrą białaczkę szpikową. Blasty ETP-OBL nie mają aberracji genetycznych powszechnie występujących w T-OBL, w tym także mutacji NOTCH1, czy delecji genu CDKN2A/B. 45, 46, 47
PODSUMOWANIE – ROLA BADAŃ MOLEKULARNYCH W DIAGNOSTYCE OBL
- Ostatnie lata okazały się bardzo owocne w zrozumieniu genetycznego podłoża patogenezy OBL. Nowe techniki molekularne umożliwiające pełną analizę całego genomu komórki pozwoliły na wykrycie zmian submikroskopowych w obrębie genów ważnych dla wzrostu i różnicowania limfocytów. Badania wykazały, że niektóre z nich mogą stanowić niezależny czynnik prognostyczny, a w przyszłości narzędzie do kwalifikacji pacjentów dla terapii celowanych.
- W świetle obecnych badań wydaje się, że zarówno klasyczne badania stosowane do tej pory w rutynowej praktyce laboratoryjnej (cytogenetyka oraz immunofenotypowanie), jak również genetyka molekularna, mają odrębne, ale komplementarne znaczenie w diagnostyce oraz ustalaniu rokowania OBL: w B-OBL stratyfikacja ryzyka dokonuje się razem z oceną wyniku badania cytogenetycznego, natomiast w T-OBL równolegle z badaniem immunofenotypu. Niezależnie w obu przypadkach genetyka molekularna umożliwia śledzenie choroby resztkowej dzięki zastosowaniu allelospecyficznych primerów dla indywidualnego łącza VDJ genów – TCR i Ig, jak również obecności genów fuzyjnych. Dalsze poszerzenie listy markerów różnicujących rokowanie w tych grupach OBL, które wiąże się z zastosowaniem innowacyjnych technik – może okazać się pomocne w praktyce klinicznej.
- 1. Jabourr E J, Faderl S, Kantarjian HM Adult Acute Lymphoblastic Leukemia. Mayo Clin Proc 2005; 80: 1517-27
- 2. Inaba H, Greaves M, Mullighan CG Acute lymphoblastic leukaemia. Lancet 2013; 381: 1943-55
- 3. WHO classification of Tumours of Hematopoietic and Lymphoid Tissue. IARC 2008, Lyon
- 4. Mullighan CG, Goorha S, Radtke I, et al Genome-wide analysis of genetic alterations in acute lymohoblastic leukemia. Nature 2007; 446: 758-64
- 5. Ribero RC, Abromowitch M, Raimondi SC, et al. Clinical and biological hallmarks of the Philadelphia chromosome in childhood acute lymphoblastic leukemia. Blood 1987; 70: 948-53
- 6. Crist W, Caroll A, Shuster J, et al. Philadelphia chromosome positive childhood acute lymphoblastic leukemia: clinical and cytogenetic characteristics and treatment outcome. A Pediatric Oncology Group study. Blood 1990; 76: 489-94
- 7. Cazzaniga G, van Delft FW, Lo Nigro L, et al. Developmantal origins and impact of BCR-ABL1 fusion and IKZF1 deletions in monozygotic twins with Ph+ acute lymphoblastic leukemia. Blood 2011; 118: 5559-64
- 8. Mullighan CG, Miller CB, Radtke I, et al. BCR-ABL1 lymphoblastic leukemia is characterized by deletions of Ikaros. Nature 2008; 453: 110-113
- 9. Den Boer M, van Slegtenhorst M, De Menezes RX, et al. A subtype of childhood acute lymphoblastic leukemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol 2009; 10: 125-34
- 10. Mullighan CG, Zhang J, Harvey RC, et al. JAK mutations in high-risk childhood acute lymphoblastic leukemia. Proc Natl Acad Sci 2009; 106: 9414-18
- 11. Harvey RC, Mullighan CG, Chen IM, et al.Rearrangement of CRLF2 is associated with mutation of JAK kinases, alteration of IKZF1, Hispanic/Latino ethnicity, and a poor outcome in pediatric B-progenitor acute lymphoblastic leukemia. Blood 2010; 115: 5312-21
- 12. Roberts KG, Morin RD, Zhang J, et al. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell 2012; 22: 153-66
- 13. Yoda A, Yoda Y, Chiaretti S, et al. Functional screening identifies CRLF2 in precursor B-cell acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 2010; 107:252-7
- 14. Notta F, Mullighan CG, Wang JC, et al. Evolution of human BCR-ABL1 lymphoblastic leukaemia-initiating cells. Nature. 2011; 469: 362-7
- 15. Feldhahn N. et al. Activation-induced cytidine deaminase acts as a mutator in BCR-ABL1-transformed acute lymphoblastic leukemia cells
- 16. Meyer C, Schneider B, Jakob S, et al. The MLL recombinome of acute leukemias. Leukemia 2006; 20: 777-84
- 17. Pieters R. Infant acute lymphoblastic leukemia: Lessons learned and future directions. Curr Hematol Malig Rep. 2009; 4: 167-74
- 18. Bueno C, Montes R, Catalina P, et al. Insights into the cellular origin and etiology of the infant pro-B acute lymphoblastic leukemia with MLL-AF4 rearrangement. Leukemia 2011; 25: 400-10
- 19. Teitell MA, Pandolfi PP Molecular genetics of acute lymphoblastic leukemia. Annu Rev Pathol 2009; 4: 175-98
- 20. Armstrong SA, Staunton JE, Silverman LB, et al. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat Genet 2002; 30: 41-7. + Stumpel DJ, Schneider P, van Roon EH, et al. Specific promoter methylation identifies different subgroups of MLL-rearranged infant acute lymphoblastic leukemia, influences clinical outcome, and provides therapeutic options. Blood 2009; 114: 5490-8
- 21. Al-Sweedan SA, Neglia JP, Steiner ME, et al. Characteristics of patients with TEL-AML1-positive acute lymphoblastic leukemia with single or multiple fusions. Pediatr Blood Cancer 2007; 48: 510-4
- 22. van Dongen JJ, Macintyre EA, Gabert JA, et al. Standardized RT-PCR analysis of fusion gene transcripts from chromosome aberrations in acute leukemia for detection of minimal residual disease. Report of the BIOMED-1 Concerted Action: investigation of minimal residual disease in acute leukemia. Leukemia 1999; 13:1901-28
- 23. Wiemels JL, Cazzaniga G, Daniotti M, et al. Prenatal origin of acute lymphoblastic leukaemia in children. Lancet 1999; 354: 1499-503
- 24. Waanders E, Scheijen B, van der Meer LT, et al. The origin and nature of tightly clustered BTG1 deletions in precursor B-cell acute lymphoblastic leukemia support a model of multiclonal evolution. PLoS Genet 2012; 8
- 25. Robinson HM1, Harrison CJ, Moorman AV, et al. Intrachromosomal amplification of chromosome 21 (iAMP21) may arise from a breakage-fusion-bridge cycle. Genes Chromosomes Cancer 2007; 46: 318-26
- 26. Attarbaschi A, Mann G, Panzer-Grümayer R, et al. Minimal residual disease values discriminate between low and high relapse risk in children with B-cell precursor acute lymphoblastic leukemia and an intrachromosomal amplification of chromosome 21: the Austrian and German acute lymphoblastic leukemia Berlin-Frankfurt-Munster (ALL-BFM) trials. Clin Oncol 2008; 26: 3046-50
- 27. Zaliova M, Zimmermannova O, Dörge P, et al. ERG deletion is associated with CD2 and attenuates the negative impact of IKZF1 deletion in childhood acute lymphoblastic leukemia. Leukemia 2014; 28:182-5
- 28. Clappier E, Auclerc MF, Rapion J, et al. An intragenic ERG deletion is a marker of an oncogenic subtype of B-cell precursor acute lymphoblastic leukemia with a favorable outcome despite frequent IKZF1 deletions. Leukemia 2014; 28: 70-7
- 29. Aifantis I, Raetz E, Buonamici S Molecular pathogenesis of T-cell leukaemia and lymphoma. Nat Rev Immunol 2008; 8: 380-90
- 30. Heerema NA, Sather HN, Sensel MG, et al. Frequency and clinical significance of cytogenetic abnormalities in pediatric T-lineage acute lymphoblastic leukemia: a report from the Children’s Cancer Group. J Clin Oncol 1998;16 :1270-8
- 31. Haydu JE, Ferrando AA Early T-cell precursor acute lymphoblastic leukaemia. Curr Opin Hematol 2013; 20: 369-73
- 32. Van Vlierberghe P, Ferrando A The molecular basis of T-cell acute lymphoblastic leukemia. J Clin Invest. 2012; 122: 3398-406
- 33. Asnafi V, Radford-Weiss I, Dastugue N, et al. CALM-AF10 is a common fusion transcript in T-ALL and is specific to the TCR gamma delta lineage. Blood 2003;102: 1000-06
- 34. Asnafi V, Buzyn A, Thomas X, et al. Impact of TCR status and genotype on outcome in adult T-cell acute lymphoblastic leukemia: a LALA-94 study. Blood 2005;105: 3072-8
- 35. Janssen JW, Ludwig WD, Sterry W, et al. SIL–TAL1 deletion in T-cell acute lymphoblastic leukemia. Leukemia 1993; 7: 1204-10
- 36. Mansur MB, Emerenciano M, Brewer L, et al. SIL-TAL1 fusion gene negative impact in T-cell acute lymphoblastic leukemia outcome. Leuk Lymphoma 2009; 50: 1318-25
- 37. Bash RO, Crist WM, Shuster JJ, et al. Clinical features and outcome of T-cell acute lymphoblastic leukemia in childhood with respect to alterations at the TAL1 locus: a Pediatric Oncology Group study. Blood 1993; 81: 2110-17
- 38. Asnafi V1, Beldjord K, Boulanger E Analysis of TCR, pT alpha, and RAG-1 in T-acute lymphoblastic leukemias improves understanding of early human T-lymphoid lineage commitment. Blood. 2003; 101:2693-703
- 39. Wang D, Zhu G, Wang N, et al. SIL-TAL1 rearrangement is related with poor outcome: a study from a Chinese institution. PLoS One 2013; 8
- 40. Graux C, Cools J, Melotte C, et al. Fusion of NUP214 to ABL1 on amplified episomes in T-cell acute lymphoblastic leukemia. Nat Genet 2004; 36: 1084-89
- 41. Ranganathan P, Weaver KL, Capobianco AJ, et al. Notch signalling in solid tumours: a little bit of everything but not all the time. Nature Rev Cancer 2011; 11: 338-351
- 42. O’Neil J, Grim J, Strack P, et al. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. J Exp Med. 2007 ; 204:1813-24
- 43. Palomero T, Ferrando A. Therapeutic targeting of NOTCH1 signaling in T-cell acute lymphoblastic leukemia. Clin Lymphoma Myeloma 2009; 9
- 44. Real PJ, Ferrando AA NOTCH inhibition and glucocorticoid therapy in T-cell acute lymphoblastic leukemia. Leukemia 2009; 23: 1374-7
- 45. Coustan-Smith E, Mullighan, CG, Onciu M, et al. Early T-cell precursor leukemia: a subtype of very high-risk acute lymphoblastic leukemia identified in two independent cohorts. Lancet Oncol 2009; 10: 147-56
- 46. Mullighan CG The molecular genetic makeup of acute lymphoblastic leukemia. Hematology Am Soc Hematol Educ Program 2012; 2012: 389-96
- 47. Neumann M, Greif PA, Baldus CD Mutational landscape of adult ETP-ALL. Oncotarget 2013; 4: 954-5
Następny artykuł:
Genetyczne zróżnicowanie ostrych białaczek limfoblastycznych


 Dodaj do ulubionych
Dodaj do ulubionych
