

Dążymy do celu
Wirus zapalenia wątroby typu C w patogenezie insulinooporności
Dr n. med. Irmina Korzeniewska-Dyl1
Dr n. med. Łukasz Kępczyński2

Dr n. med. Irmina Korzeniewska-Dyl

Dr n. med. Łukasz Kępczyński
Wirus zapalenia wątroby typu C (HCV) jest należącym do rodziny flaviviridae wirusem RNA, który wywołuje przewlekłe zapalenie wątroby u około 160 mln osób na świecie.[1] Dane epidemiologiczne wskazują na liczne powiązania zaburzeń metabolicznych, takich jak insulinooporność oraz stłuszczenie wątroby, z zakażeniem HCV.
W 1998 roku po raz pierwszy pojawiły się doniesienia o zwiększonej (4-8-krotnie) częstości występowania nowych przypadków cukrzycy u chorych HCV dodatnich.[2] Upośledzoną tolerancję glukozy i cukrzycę spotyka się częściej u chorych zakażonych HCV w porównaniu z chorymi zakażonymi HBV.[3] Obserwowano, że infekcja HCV wyprzedza wystąpienie cukrzycy u chorych w starszym wieku, z nadwagą i u płci męskiej, a insulinooporność mierzona za pomocą wskaźnika HOMA-IR lub za pomocą euglikemicznej hiperinsulinowej klamry metabolicznej jest zwiększona u osób zakażonych różnymi typami HCV w porównaniu z resztą populacji.[4]
Metaanaliza zbierająca dane na temat cukrzycy w zakażeniu HCV potwierdziła sprawstwo wirusa w rozwoju insulinooporności.[5] Jednocześnie obserwowano, że eradykacja HCV prowadzi do poprawy wrażliwości na insulinę.[6]
Interferencja HCV w szlak sygnału insuliny
W świetle obecnych danych zakłada się bezpośrednią ingerencję białek rdzeniowych wirusa w wewnątrzkomórkowy szlak sygnału insuliny w hepatocycie. Istotą działania insuliny po połączeniu z receptorem insulinowym (IR) jest przemieszczenie się transportera glukozy GLUT-4 na powierzchnię komórek, co umożliwia dokomórkowy transport glukozy. Efekt działania insuliny zależy od złożonej kaskady powiązanych ze sobą reakcji enzymatycznych. Po połączeniu insuliny z receptorem dochodzi do autofosforylacji receptora, a następnie do fosforylacji tyrozynowych reszt wewnątrzkomórkowo położonych białek adaptorowych – substratów receptora insulinowego: IRS-1 i IRS-2.
W przeciwieństwie do fosforylacji reszt tyrozynowych fosforylacja reszt serynowych osłabia sygnał insulinowy – jest to rodzaj negatywnej regulacji działania insuliny w warunkach fizjologicznych, która jednak w pewnych warunkach może przełożyć się na rozwój insulinooporności.
Inne czynniki, które hamują aktywację białek IRS, to: fosfatazy białkowe tyrozyny (PTPs), szczególnie PTP1B, która defosforyluje reszty tyrozynowe receptora insulinowego lub IRS-1/2, a także supresory sygnału cytokinowego (SOCS), takie jak SOCS-1 i SOCS-3, które prowadzą do degradacji IRS-1/2 zależnej od ubikwityny.[7]
Kluczowy dla metabolicznego efektu insuliny jest szlak kinazy fosfatydyloinozytolu-3 (PI3K) i kinazy białkowej B – inaczej kinazy Akt. Fosforylacja reszty tyrozyny w IRS-1 prowadzi do aktywacji kinazy fosfatydyloinozytolu-3 (PI3K) głównie klasy I. Kinaza PI3 klasy I fosforyluje difosfatydyloinozytol do trifosforanu fosfatydyloinozytolu (PIP3), który stymuluje fosforylację i aktywację kinazy Akt. Czynniki zmniejszające ilościowy stosunek PIP3 do PIP2 hamują aktywację kinazy Akt, która jest najistotniejsza dla translokacji GLUT-4 na powierzchnię komórek.
Kinaza Akt po aktywacji hamuje kinazę 3-syntazy glikogenu 3 i aktywuje fosfatazę białkową 1 (PP1), co prowadzi do uczynnienia syntazy glikogenu (GS). Insulina hamuje zatem glukoneonezę i glikogenolizę. Ten sam efekt insulina wywołuje również poprzez wpływ na różne czynniki transkrypcyjne, takie jak wątrobowy czynnik jądrowy 4 (HNF-4), czynniki należące do rodziny FoxO oraz na koaktywator 1 receptora PPR-γ. Insulina ponadto nasila lipogenezę przez wpływ na białko SREBP-1 (sterol regulatory element binding protein 1).
Poprzez szlak PI3K oraz szlak mTOR (mammalian target of rapamycin) oraz alternatywny szlak zależny od kinaz aktywowanych miogenem (rodzina MAPK) insulina pobudza syntezę białek, a także stymuluje proliferację i różnicowanie komórek.[8]
W modelach eksperymentalnych wykazano, że HCV zaburza szlak sygnału insulinowego w hepatocytach w różnych mechanizmach postreceptorowych. Głównym celem HCV wydaje się szlak kinazy Akt, której fosforylacja jest upośledzona mimo stymulacji insuliną.[4]
W komórkach zakażonych HCV zwiększa się znacząco fosforylacja serynowa IRS-1, a zmniejsza się fosforylacja tyrozynowa, co skutkuje osłabieniem kaskady prowadzącej do aktywacji kinazy Akt.[7]
Wzrasta ekspresja genów glukoneogenezy
Istotną rolę w rozwoju insulinooporności w zakażeniu HCV odgrywa rodzina supresorów sygnału cytokinowego, szczególnie SOCS-3 i SOCS-7, których ekspresja rośnie w zakażonych komórkach. SOCS hamują kaskadę sygnału insuliny przez kierowanie białek IRS1/2 na drogę ubikwitynozależnej degradacji proteosomalnej.[9]
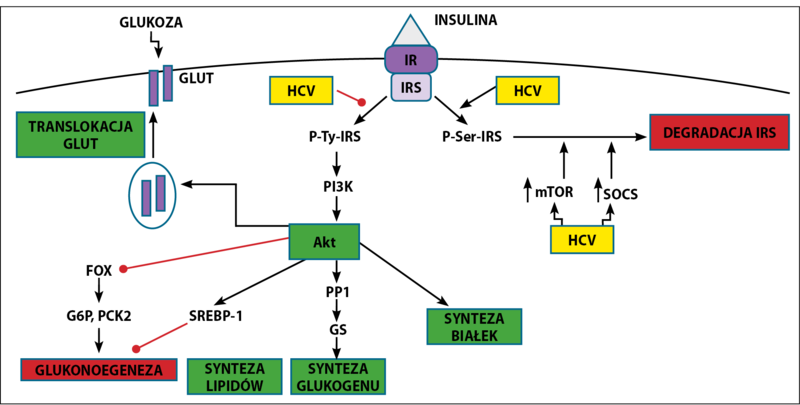
Ryc. 1. Wpływ wirusa zapalenia wątroby typu C na szlak sygnału insuliny w hepatocycie (opracowanie własne na podst.[7]).
W modulowaniu aktywności IRS-1 dużą rolę odgrywa pobudzenie szlaku mTOR, które obserwuje się w komórkach zakażonych HCV. Wirus znacząco zwiększa fosforylację seryny w pozycji 1101 w IRS-1, a tak fosforylowane formy IRS-1, dzięki działaniu szlaku mTOR, są uwalniane z wewnątrzkomórkowych kompleksów, co umożliwia ich degradację.
Ponadto zakażenie HCV nasila ekspresję genów glukoneogenezy, takich jak geny dla glukozo-6-fosfatazy (G6P) oraz karboksykinazy 2 fosfoeonolopirogronianu (PCK2), prowadząc do zwiększonej produkcji glukozy. HCV zmniejsza ekspresję receptorów PPAR-γ (peroxisomeproliferator-activated receptor), a także ekspresję genu SLC2A4, kodującego transporter GLUT-4, koniecznego do wychwytu glukozy. Wzrost produkcji glukozy oraz osłabienie jej wychwytu przez tkanki skutkuje rozwojem insulinooporności.[10]
Artykuł ukazał się

Czasopismo
Diabetologia po Dyplomie
Prenumerata

Stany Nagłe po Dyplomie
Nr 02 (kwiecień) / 2026
Nowe publikacje

Książka
Stany nagłe niemedyczne
19,90 zł
Wideo
