Sympozjum: immunologia kliniczna
Czy u pacjenta występuje zespół nawracającej gorączki?
S. Savic P. Wood
Zespoły nawracającej gorączki to schorzenia należące do większej grupy chorób, zwanych zespołami autoimmunologicznymi. Po odkryciu podłoża genetycznego zespołu nawracającej gorączki związanego z receptorem TNF (TRAPS – tumour necrosis factor receptor-associated periodic fever syndrome), po raz pierwszy w 1999 r. zaproponowano termin autozapalenie, aby odróżnić immunopatogenetycznie zespół nawracającej gorączki (PFS – periodic fever syndrome) od klasycznych postaci chorób autoimmunologicznych.1
Termin „autozapalenie” został później poszerzony i obejmuje wiele chorób o podłożu immunologicznym, które rozwijają się na skutek patologicznego stanu zapalnego wywołanego w głównej mierze przez wrodzone mechanizmy odporności nieswoistej. Stan ten różni się od chorób o podłożu autoimmunologicznym, w których proces chorobowy jest przeważnie spowodowany nieprawidłową odpowiedzią adaptacyjną układu immunologicznego i uczestniczą w nim limfocyty T i B rozpoznające swoiście antygeny. Zrozumienie, że proces zapalny może być spowodowany przez oddzielne mechanizmy układu immunologicznego – albo wrodzone (nieswoiste), albo nabyte – doprowadziło do ponownej klasyfikacji chorób autoimmunologicznych i wyodrębnienia chorób autozapalnych, chociaż przyznaje się, że wiele chorób o pośrednim fenotypie w znacznym stopniu nakłada się na siebie.2 Odkrycie genetycznego podłoża różnych zespołów nawracającej gorączki spowodowało zmianę podejścia do chorób zapalnych wywołanych procesami immunologicznymi i pozwoliło na lepsze poznanie chorób zapalnych, w których powstawaniu decydującą rolę odgrywają nieprawidłowości wrodzonego układu immunologicznego.
W przedstawionym artykule zwrócono uwagę na swoiste kliniczno-patologiczne cechy zespołów nawracającej gorączki, aby ułatwić ich rozpoznawanie w codziennej praktyce klinicznej.
Genetyka i immunopatologia zespołów nawracającej gorączki
Określenie PFS odnosi się zwykle do grupy gorączek uwarunkowanych genetycznie, występujących okresowo, które są dziedziczone w sposób albo autosomalny recesywny, albo dominujący. Niektóre objawy kliniczne PFS są takie same jak objawy innych dziedzicznych zespołów autozapalnych wymienionych w tabeli 1. Najczęstszą i najlepiej poznaną jest rodzinna gorączka śródziemnomorska (FMF – familial Mediterranean fever), pierwsza zidentyfikowana jednostka PFS o etiologii genetycznej (mutacja genu MEVF), wykrytej w 1997 r.3,4 Etiologię genetyczną dwóch kolejnych PFS poznano w 1999 r:
- TRAPS (wcześniej znany jako rodzinna gorączka irlandzka [familial Hibernian fever]) jest wywołany mutacją genu TNFRSF1a1
- zespół zwiększonego stężenia IgD (HIDS – hyperimmunoglobulinaemia D with periodic fever syndrome) jest spowodowany homozygotyczną mutacją genu MVK.5,6
Kluczowe zagadnienia
- Zespoły nawracającej gorączki występują bardzo rzadko i są spowodowane zaburzeniami regulacji mechanizmów odporności nieswoistej.
- Większość zespołów nawracającej gorączki pojawia się w dzieciństwie. Charakteryzują się one samoistnymi epizodami gorączki, którym towarzyszy uogólniony i miejscowy stan zapalny.
- Amyloidoza AA jest ciężkim długotrwałym powikłaniem występującym u pewnego odsetka pacjentów z zespołem nawracającej gorączki.
- Rozpoznanie zespołu nawracającej gorączki stanowi wyzwanie dla lekarza i polega m.in. na wykluczeniu innych częstszych stanów chorobowych, np. utajonych ognisk zakażenia, chorób autoimmunizacyjnych i nowotworów.
- Dostępne są rutynowe badania genetyczne służące rozpoznawaniu zespołów nawracającej gorączki.
- Niewyjaśniona amyloidoza AA wymaga wykluczenia zespołu nawracającej gorączki.
- W leczeniu zespołów nawracającej gorączki przydatne są celowane terapie przeciwcytokinowe.
W 2001 r. odkryto, że za występowanie zespołu objawów zapalnych po ekspozycji na zimno (FCAS – familial cold autoinflammatory syndrome) oraz zespołu Muckle’a-Wellsa (MWS – Muckle-Wells syndrome) jest odpowiedzialna heterozygotyczna mutacja genu CISA1.7,8 Rok później w tym samym genie wykryto kolejną mutację, która odpowiada za powstawanie noworodkowej zapalnej choroby wieloukładowej (NOMID – neonatal onset multisystem inflammatory disease), znanej również jako przewlekły neurologiczno-skórno-stawowy zespół występujący u niemowląt (CINCA – chronic infantile neurologic, cutaneous and articular syndrome).9 Później wprowadzono szersze pojęcie – zespołu nawracającej gorączki związanego z kriopiryną (CAPS – cryopyrin associated periodic fever syndrome) – które odnosi się do zespołów klinicznych o wspólnym podłożu genetycznym. W 2008 r. opisano pierwsze przypadki nawracającej gorączki przypominającej FCAS, lecz związanej z mutacją NLRP12.10
Stosunkowo niedawno opisano niedobór antagonistów receptora interleukiny 1 (DIRA – deficiency of interleukin-1 receptor antagonists). To zaburzenie różni się pod względem obrazu klinicznego od CAPS, lecz ma podobną etiologię immunopatologiczną – u jego podłoża leżą zaburzenia na drodze przekazywania sygnału IL-1.11 DIRA wywołany jest homozygotyczną mutacją genu IL1RN i należy do grupy autozapalnych chorób przebiegających z ropieniem.
Wspólną cechą wszystkich zespołów nawracającej gorączki są mutacje genetyczne, które w różnym stopniu wpływają na wytwarzanie ważnych cytokin prozapalnych – TNF i IL-1 – wydzielanych przede wszystkim przez komórki wrodzonego układu immunologicznego (tab. 1).
|
Tabela 1. Wrodzone zespoły autozapalne: cechy genetyczne i immunopatologiczne |
||||
|
Typ gorączki |
Dziedziczenie |
Gen |
Białko |
Immunopatologia |
|
FMF |
AR |
MEVF |
Piryna |
Zmniejszona apoptoza makrofagów, zahamowanie ujemnego sprzężenia zwrotnego wytwarzania IL-1 |
|
HIDS |
AR |
MVK |
MVK |
Niejasna |
|
TRAPS |
AD |
TNFRSF1A |
TNFRSF1A |
Zmniejszone złuszczanie receptora, wadliwa apoptoza indukowana TNF, stałe (konstytucyjne) przesyłanie sygnału NF-κB, upośledzony transport receptorów |
|
CAPS: FCAS MWS CINCA/NOMID |
AD W większości de novo |
CIAS1 |
Kriopiryna (NLRP3) |
Zwiększone samoistne wytwarzanie i uwalnianie IL-1 |
|
NAPS12 |
AD |
NLRP12 |
NLRP12 |
Zwiększone wytwarzanie IL-1β |
|
Inne wrodzone zespoły autozapalne |
||||
|
DIRA |
AR |
IL1RN |
Niedobór antagonisty receptora IL-1 |
Antagonista receptora IL-1 |
|
PAPA |
AR |
IPSTPIP1 |
Fosfataza białkowa 1 oddziałująca z proliną, seryną, treoniną |
Zaburzona reorganizacja aktyny powodująca nieprawidłową odpowiedź zapalną |
|
CRMO |
AR |
LPIN2 |
Lipina 2 |
Niejasny |
|
Zespół Blaua |
AD |
CARD15 |
NOD2 |
Zwiększona aktywacja NF-κB |
AD (autosomal dominant inheritance) – dziedziczenie autosomalne dominujące; AR (autosomal recessive inheritance) – dziedziczenie autosomalne recesywne; CAPS (cryopyrin associated periodic fever syndrome) – zespół nawracającej gorączki związany z kriopiryną; CIAS (cold-induced autoinflammatory syndrome) – gen zespołu autozapalnego wywołanego zimnem; CINCA (chronic infantile neurologic, cutaneous and articular syndrome) – przewlekły neurologiczno-skórno stawowy zespół występujący u niemowląt; CRMO (chronic recurrent multifocal osteomyelitis and congenital dyserythropoietic anaemia) – przewlekłe nawracające wieloogniskowe zapalenie kości i szpiku oraz wrodzona niedokrwistość dyserytropoetyczna; DIRA (deficiency of interleukin-1 receptor antagonists) – niedobór antagonistów receptora interleukiny 1; FMF (familial Mediterranean fever) – rodzinna gorączka śródziemnomorska; HIDS (hyperimmunoglobulinaemia D with period fever syndrome) – zespół zwiększonego stężenia IgD; MVK (mevalonate kinase) – kinaza mewalonianowa; MWS (Muckle-Wells syndrome) – zespół Muckle’a-Wellsa; NAPS12 (NLRP12-associated periodic syndrome) – zespół nawracającej gorączki spowodowany występowaniem genu NLRP12; NF-κB (nuclear factor κB) – czynnik jądrowy κB; NLRP3 (nucleotide-binding domain, leucine-rich repeat containing protein [NLR] family, pyrin domain containing 3) – receptory NOD-podobne zawierające domenę pirynową 3; NLRP12 – gen zespołu nawracającej gorączki; NOD (nucleotide-binding oligomerisation) – białko odgrywające rolę w regulacji układu odpornościowego; NOMID (neonatal onset multisystem inflammatory disease) – noworodkowa zapalna choroba wieloukładowa; PAPA (pyogenic sterile arthritis, pyoderma gangrenosum, acne) – zapalenie stawów, piodermia zgorzelinowa, trądzik; TNF (tumour necrosis factor) – czynnik martwicy nowotworów; TNFRSF1A (tumour necrosis factor receptor superfamily member 1A) – białko/gen rodziny receptorów dla czynnika martwicy nowotworów 1A; TRAPS (tumour necrosis factor receptor-associated periodic fever syndrome) – zespół nawracającej gorączki związany z receptorem TNF
Teorię tę potwierdza nie tylko fakt, że zmutowane geny bezpośrednio uczestniczą w procesie produkcji IL-1 i TNF, ale również obserwacja, że w tych stanach chorobowych korzystne rezultaty przynoszą terapie blokujące działanie tych dwóch cytokin (ryc.).12-14
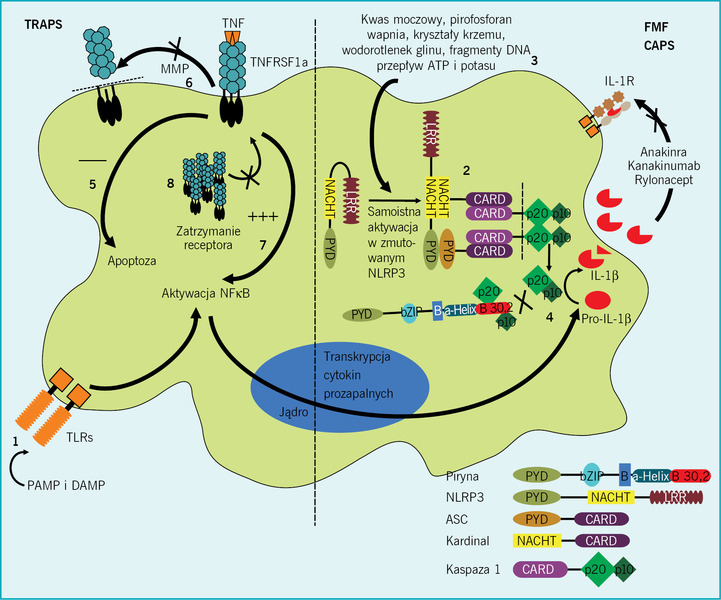
Rycina. Immunologia CAPS i TRAPS
Objawy kliniczne
Opis przypadku
P.W., 43-letni mężczyzna rasy kaukaskiej, do późnego wieku młodzieńczego zdrowy. Po tym okresie wystąpiły u niego epizodyczne napady, charakteryzujące się początkowo bólami brzucha i bólami w klatce piersiowej, którym towarzyszyła gorączka i podwyższone stężenia wskaźników zapalenia. Napady te pojawiały się bez uchwytnej przyczyny i ustępowały samoistnie w ciągu 7-10 dni. Na przestrzeni lat wykonano u niego wiele badań, aby wykluczyć stan zapalny, nowotwór i choroby autoimmunizacyjne; niektóre z nich przeprowadzano wielokrotnie (np. tomografię komputerową klatki piersiowej i jamy brzusznej). Przeprowadzono także appendektomię i laparoskopię zwiadowczą, jednak żaden z tych zabiegów nie pozwolił na ustalenie rozpoznania.
Do pierwotnego problemu zdrowotnego dołączyła się wędrująca wysypka rumieniowata, której towarzyszył ból mięśni. Wypróbowano różne schematy leczenia empirycznego. Jedynym skutecznym sposobem okazało się stosowanie glikokortykosteroidów ogólnoustrojowych, np. prednizolonu, które skracało czas trwania napadów, kiedy leczenie rozpoczynano w momencie występowania typowych objawów. Przez ponad 20 lat u pacjenta nie rozpoznano zespołu nawracającej gorączki związanego z receptorem TNF, czyli TRAPS. Jedynym członkiem rodziny, u którego występowały podobne objawy, była jego córka; objawy kliniczne wystąpiły u niej w podobnym wieku jak u ojca.
Opisany przypadek ilustruje niektóre z klasycznych objawów klinicznych występujących w zespole nawracającej gorączki, jak również problemy diagnostyczne, jakie mogą pojawić się w takich przypadkach. Zwykle występują epizodyczne samorzutne napady uogólnionego zapalenia, którym towarzyszy gorączka, zapalenie surowicze i różnego stopnia zajęcie mięśni, stawów, układu nerwowego i skóry, w zależności od typu zespołu nawracającej gorączki (tab. 2).15,16
|
Tabela 2. Objawy kliniczne zespołów nawracającej gorączki |
|||||||
|
Zespół nawracającej gorączki |
Czas trwania napadu |
Objawy skórne |
Objawy mięśniowo- |
Objawy brzuszne |
Objawy oczne |
Inne |
Amyloidoza |
|
FMF |
1-3 dni |
Różopodobny |
Epizodyczne zapalenie jednego stawu, zapalenie stawów krzyżowo-biodrowych |
Jałowe zapalenie otrzewnej (85%) |
Rzadko |
Zapalenie opłucnej, osierdzia, ból/obrzęk moszny |
Ryzyko jest różne w zależności od genotypu i czynników środowiskowych |
|
HIDS |
3-7 dni |
Rumieniowate |
Bóle stawów, nienadżerkowe zapalenie stawów |
Ból, wymioty, biegunka, rzadko zapalenie otrzewnej |
Rzadko |
Podczas napadów limfadenopatia szyjna, owrzodzenia aftowe, splenomegalia |
Wyjątkowo rzadkie |
|
TRAPS |
7-21 dni, czasami dłużej |
Uogólniona wędrująca wysypka różopodobna nakładająca się |
Silne wędrujące bóle mięśniowe, bóle stawów, nienadżerkowe zapalenie jednego stawu |
Ból, zapalenie otrzewnej, biegunka, zaparcia |
Zapalenie spojówek, obrzęk w okolicy oczodołowej |
Zapalenie opłucnej, bóle głowy |
10% |
|
CAPS: |
|||||||
|
FCAS |
<24 h |
Uogólniona wysypka pokrzywkowa |
Bóle stawów |
Zapalenie spojówek |
Bóle mięśni, głowy, senność |
Rzadko |
|
|
MWS |
24-48 h |
Bóle stawów, zapalenie kilku stawów |
Zapalenie spojówek, zapalenie nadtwardówki |
Głuchota czuciowo-nerwowa |
25% |
||
|
CINCA/NOMID |
Stale z zaostrze- |
Pokrzywka, grudki/blaszki rumieniowe i obrzękowe |
Nadmierny rozrost nasad kości ze zniekształceniem stawów |
Rzadko |
Zapalenie błony naczyniowej przedniego odcinka oka, zmiany w obrębie tarczy nerwu wzrokowego, ślepota |
Jałowe zapalenie opon mózgowo-rdzeniowych, opóźnienie rozwoju umysłowego, głuchota, niskorosłość, nadmiernie rozwinięte guzy czołowe (frontal bossing), opóźnione zarastanie ciemiączek, nos siodełkowaty, pałeczkowatość palców |
Możliwa w wieku dorosłym |
|
NAPS12 |
3-5 dni |
Pokrzywka |
Bóle stawów, mięśni |
Żadne |
Wrażliwość na zimno |
Nieznana |
|
CINCA (chronic infantile neurologic, cutaneous and articular syndrome) – przewlekły neurologiczno-skórno-stawowy zespół występujący u niemowląt; FCAS (familial cold autoinflammatory syndrome) – zespół objawów zapalnych po ekspozycji na zimno; FMF (familial Mediterranean fever) – rodzinna gorączka śródziemnomorska; HIDS (hyperimmunoglobulinaemia D with period fever syndrome) – zespół zwiększonego stężenia IgD;
MWS – (Muckle-Wells syndrome) – zespół Muckle’a-Wellsa; NAPS12 (NLRP12-associated periodic syndrome) – zespół nawracającej gorączki spowodowany występowaniem genu NLRP12;NOMID (neonatal onset multisystem inflammatory disease) – noworodkowa zapalna choroba wieloukładowa; TRAPS (tumour necrosis factor receptor-associated periodic fever syndrome) – zespół nawracającej gorączki związany z receptorem TNF
Większość tych chorób ujawnia się już w dzieciństwie, lecz czasami, tak jak w przedstawionym przypadku, pierwsze wyraźne objawy mogą pojawić się we wczesnym okresie dorosłości. W klasycznym obrazie klinicznym napady gorączki mają charakter epizodyczny, występują naprzemiennie z okresami całkowicie wolnymi od objawów. Jednak niektóre postaci, np. ciężkie zespoły CAPS mają charakter bardziej przewlekły: w FMF i w TRAPS między okresami objawowymi może utrzymywać się subkliniczny stan zapalny.17