III Kongres Akademii po Dyplomie Okulistyka już w ten piątek! Kup bilet i dołącz do ekspertów podczas Siatkówka Meeting! Sprawdź >
Program edukacyjny: hipertensjologia
Inhibitory reniny – nowe leki w terapii nadciśnienia tętniczego
Helmy M. Siragy, MD
W SKRÓCIE
Renina to enzym, który katalizuje pierwszą – kontrolującą tempo pozostałych – reakcję przekształcenia angiotensynogenu w angiotensynę I, a przez to wpływa na wytwarzanie angiotensyny II. Hamowanie aktywności układu renina-angiotensyna (RAS) wykorzystuje się w leczeniu wielu chorób układu krążenia. Zapewnienie odpowiedniej kontroli wciąż jednak stwarza trudności i wymaga jednoczesnego podawania kilku leków. Stosowanie inhibitorów RAS nie powstrzymuje całkowicie wytwarzania angiotensyny II, zatem wciąż może ona przyczyniać się do rozwoju powikłań narządowych. Już od 20 lat kuszącą metodą całkowitego blokowania RAS wydaje się hamowanie aktywności reniny. Niedawno dopuszczono do stosowania aliskiren, pierwszy lek należący do nowej klasy środków przeciwnadciśnieniowych – inhibitorów reniny. W badaniach klinicznych wykazano, że lek ten stosowany w monoterapii lub w skojarzeniu z hydrochlorotiazydem, inhibitorami ACE lub sartanami znacząco obniża ciśnienie tętnicze. Trwają badania oceniające przydatność inhibitorów reniny w terapii chorób nerek i serca.
Wprowadzenie

Rycina 1. Schemat przedstawiający układ renina-angiotensyna, czynniki pobudzające wytwarzanie i aktywację reniny oraz blokujące receptory AT1 (krótka pętla ujemnego sprzężenia zwrotnego)
Renina, należąca do grupy proteaz asparaginianowych, wytwarzana jest głównie przez komórki przykłębuszkowe w nerkach w postaci nieaktywnej, określanej mianem proreniny. Jej wydzielanie wzrasta pod wpływem stymulacji przez czynniki, jak: małe spożycie sodu, hipowolemia, hipokaliemia, zwiększona aktywność współczulnego układu nerwowego (głównie w wyniku aktywacji receptorów β-adrenergicznych) oraz podwyższone stężenie prostaglandyny E2 i I2. Przekształcenie proreniny w reninę (aktywną postać) zachodzi pod wpływem obniżonego pH, niskiej temperatury i enzymów, takich jak kalikreina, trypsyna, plazmina i katepsyna D.1 Cząsteczka reniny składa się z dwóch homologicznych płatów rozdzielonych szczeliną, w której znajduje się miejsce aktywne katalitycznie. Jedyną znaną formą aktywności proteolitycznej tego enzymu jest wytwarzanie angiotensyny I z prekursora – angiotensynogenu, co w konsekwencji prowadzi do wzrostu wydzielania angiotensyny II, która ma właściwości naczynioruchowe. W normalnych warunkach zwiększone wytwarzanie angiotensyny II poprzez stymulację receptorów typu 1 dla angiotensyny hamuje wydzielanie reniny, tworząc krótką pętlę ujemnego sprzężenia zwrotnego (ryc. 1).
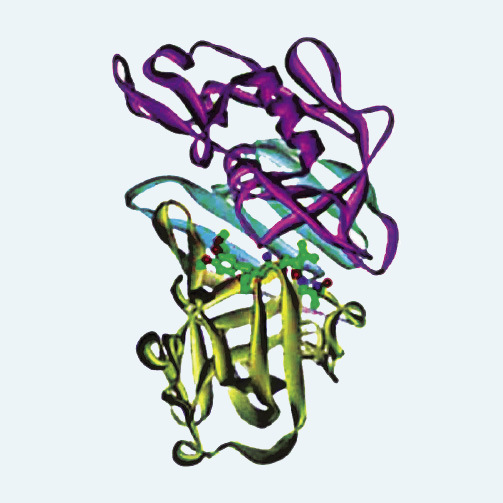
Rycina 2. Struktura molekularna ludzkiej reniny
W układzie krążenia występuje przede wszystkim aktywna renina. Czasem jednak stwierdza się podwyższone stężenie proreniny we krwi, np. u chorych na cukrzycę2 z powikłaniami mikronaczyniowymi. Renina ma złożoną strukturę. W skład cząsteczki wchodzi duża hydrofobowa nisza z dodatkowym zagłębieniem S3sp (ryc. 2) i ruchomym fałdem przykrywającym szczelinę zawierającą miejsce aktywne katalitycznie. Wyniki pierwszych badań wskazywały, że podwyższone stężenie reniny jest powiązane z ryzykiem incydentów sercowo-naczyniowych.3 U chorych z nadciśnieniem tętniczym i z podwyższonym osoczowym stężeniem tego enzymu obserwowano niezależnie od występowania innych czynników ryzyka sercowo-naczyniowego zwiększone ryzyko zawału serca. Przyjmuje się, że wyższe stężenie reniny sprzyja wytwarzaniu angiotensyny II, która odgrywa kluczową rolę w utrzymaniu homeostazy płynowej i elektrolitowej. Jest nie tylko silnym czynnikiem naczynioskurczowym, ale zwiększając agregację płytek i wytwarzanie inhibitora-1 aktywatora plazminogenu, uczestniczy też w krzepnięciu krwi, wpływa ponadto na procesy zapalne i wytwarzanie macierzy zewnątrzkomórkowej, pobudza współczulny układ nerwowy, indukuje dysfunkcję śródbłonka, stres oksydacyjny oraz uwalnianie hormonów wpływających na aktywność naczyń, takich jak aldosteron i endotelina 1. Wykazano, że inaktywacja enzymu konwertującego angiotensynę powoduje początkowo zmniejszenie wytwarzania angiotensyny II, później dochodzi jednak do zjawiska odbicia, czyli wzrostu jej stężenia mimo ciągłego stosowania inhibitorów ACE. Dzieje się tak dlatego, że istnieje alternatywny, niezależny od enzymu konwertującego angiotensynę szlak wytwarzania angiotensyny II, katalizowany przez chymazę. Również antagoniści receptorów dla angiotensyny wywołują kompensacyjny wzrost stężenia angiotensyny II. Może to tłumaczyć, dlaczego wspomniane leki tylko spowalniają rozwój powikłań narządowych, a nie eliminują ich całkowicie. Bezpośrednie inhibitory reniny umożliwiają najpełniejsze zablokowanie aktywności RAS i stwarzają szansę dalszego zmniejszenia częstości powikłań oraz zgonów sercowo-naczyniowych.
Historia bezpośrednich inhibitorów reniny

Rycina 3. Budowa chemiczna niektórych bezpośrednich inhibitorów reniny
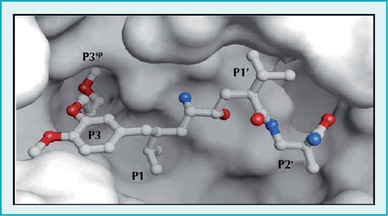
Rycina 4. Struktura krystaliczna kompleksu renina-aliskiren
Aktywność enzymatyczna reniny jest swoista gatunkowo, ludzki enzym nie przekształca bowiem angiotensyny I w angiotensynę II u szczurów. Utrudnia to badania nad ludzką reniną, które można prowadzić tylko na naczelnych oraz szczurach transgenicznych, z genami ludzkiej reniny i angiotensynogenu. O znaczeniu reniny w rozwoju nadciśnienia tętniczego świadczy to, że podanie przeciwciał swoistych dla tego enzymu wywołuje spadek ciśnienia.4 Pierwsze inhibitory reniny5-8 skutecznie hamowały jej aktywność i zmniejszały ciśnienie tętnicze.9 Główną przeszkodę w dalszym rozwoju tych leków stanowił jednak fakt, że były to peptydy, co uniemożliwiało ich doustne stosowanie i ograniczało możliwości wykorzystania. W ostatnim dziesięcioleciu opracowano doustne leki hamujące aktywność reniny.5,10 Charakteryzują się one jednak niską biodostępnością, są słabo wchłaniane z przewodu pokarmowego, mają słabe działanie hipotensyjne i krótki czas półtrwania, co zmniejsza ich atrakcyjność (ryc. 3). Na początku 2007 r. w USA jeden z takich inhibitorów reniny – aliskiren – jako pierwszy przedstawiciel grupy został dopuszczony do stosowania w terapii nadciśnienia tętniczego. W sposób wysoce wybiórczy wiąże się on ściśle z miejscem aktywnym katalitycznie w cząsteczce ludzkiej reniny (ryc. 4),11 hamując aktywność enzymu.5,10 Jest bardzo silnym inhibitorem nerkowej reniny o IC50 0,6 nM, o osoczowym czasie półtrwania od 20 h do 45 h. Maksymalne stężenie leku we krwi występuje w 3-6 h po doustnym zażyciu, a biodostępność po podaniu tą drogą szacuje się na 2,7%.5 Stężenie aliskirenu w osoczu stabilizuje się po 5-8 dniach od rozpoczęcia leczenia, nie wpływa na nie aktywność enzymów układu CYP ani warfaryna.12 Lek wydalany jest z żółcią.
Badania doświadczalne i kliniczne
W badaniu na szczurach transgenicznych z ekspresją ludzkiej reniny i angiotensynogenu aliskiren obniżał ciśnienie tętnicze krwi i zmniejszał albuminurię oraz stopień uszkodzenia serca.13 U małp pazurkowcowatych lek ten obniżał ciśnienie tętnicze o ok. 30 mmHg,10 silniej niż inhibitory ACE i sartany. U zdrowych ochotników aliskiren znacząco obniżył aktywność reninową osocza, ciśnienie tętnicze krwi i wydalanie aldosteronu z moczem.5 W grupie pacjentów z nadciśnieniem lek ten, stosowany w dawce 150, 300 i 600 mg raz dziennie, obniżał ciśnienie zależnie do dawki i był tolerowany tak samo jak placebo.14,15 W połączeniu z hydrochlorotiazydem,16 inhibitorem ACE17 lub antagonistą receptora AT1 – walsartanem,18,19 aliskiren miał silniejsze działanie hipotensyjne niż stosowany w monoterapii. W grupie otyłych chorych z nadciśnieniem tętniczym skojarzenie aliskirenu z hydrochlorotiazydem pozwalało skutecznie zredukować ciśnienie u osób, u których monoterapia tiazydowym lekiem moczopędnym była niewystarczająca.20
Wnioski
Trwają badania oceniające skuteczność aliskirenu w terapii chorób nerek i serca.21 Celem badania Aliskiren in Left Ventricular Hypertrophy (ALLAY) jest porównanie monoterapii aliskirenem, losartanem oraz leczenie skojarzone tymi dwoma lekami u chorych na nadciśnienie tętnicze krwi z przerostem lewej komory. Głównym parametrem oceny końcowej w tej próbie jest stopień przerostu i geometria komory. Projekt badania Aliskiren Observation of Heart Failure Treatment (ALOFT) zakłada porównanie efektów stosowania aliskirenu lub placebo w uzupełnieniu zoptymalizowanej standardowej terapii niewydolności serca. Za główny parametr oceny końcowej uznano w nim zmniejszenie osoczowego stężenia peptydu natriuretycznego typu B. Celem badania Aliskiren in the Evaluation of Proteinuria in Diabetes (AVOID) jest ocena aliskirenu (w porównaniu z placebo) jako uzupełnienie leczenia losartanem, zaś ocenianym parametrem jest stosunek stężenia albuminy i kreatyniny w moczu.
Komentarz
lek. Aleksander Prejbisz i prof. dr hab. med. Andrzej Januszewicz, Klinika Nadciśnienia Tętniczego, Instytut Kardiologii, Warszawa
lek. Aleksander Prejbisz
prof. dr hab. med. Andrzej Januszewicz
Artykuł Helmy’ego M. Siragy’ego w zwięzły sposób opisuje mechanizm działania nowej klasy leków hipotensyjnych – inhibitorów reniny. Przedstawia również miejsce reniny w patofizjologii chorób sercowo-naczyniowych. Omawiane zagadnienie ma szczególne znaczenie kliniczne ze względu na wprowadzenie w ostatnim czasie do stosowania klinicznego pierwszego przedstawiciela tej grupy leków – aliskirenu. W artykule przedstawiono aliskiren jako skuteczny i dobrze tolerowany lek hipotensyjny w terapii nadciśnienia tętniczego.
Warto wspomnieć o międzynarodowym wieloośrodkowym, randomizowanym badaniu prowadzonym metodą podwójnie ślepej próby z zastosowaniem placebo, którego wyniki opublikowano w 2007 r. na łamach czasopisma „Lancet”. Badaniem objęto 1797 chorych na nadciśnienie tętnicze. Wykazano podobną skuteczność hipotensyjną aliskirenu i walsartanu. Leki te stosowane w terapii skojarzonej wykazywały silniejsze działanie hipotensyjne niż każdy z nich stosowany w monoterapii. Częstość występowania zdarzeń niepożądanych była podobna w grupie placebo, w grupach otrzymujących porównywane leki w monoterapii i w grupie stosującej leczenie skojarzone za pomocą walsartanu i aliskirenu.1
Na odnotowanie zasługują trwające lub niedawno zakończone badania z zastosowaniem aliskirenu oceniające wpływ tego leku na rozwój i regresję powikłań narządowych oraz na częstość zdarzeń sercowo-naczyniowych.
Badanie Aliskiren in Left Ventricular Hypertrophy (ALLAY) miało na celu ocenę wpływu aliskirenu na przerost lewej komory u chorych na nadciśnienie tętnicze ze współistniejącym przerostem lewej komory i wskaźnikiem masy ciała >25 kg/m2. Ocena masy i geometrii lewej komory opierała się na badaniu przy użyciu rezonansu magnetycznego. Okres obserwacji wynosił 36 tygodni. Wyniki tego badania ogłoszono na sympozjum American College of Cardiology w marcu 2008 r. i opublikowano później w czasopiśmie „Circulation”. Wykazano, że aliskiren i losartan w podobnym stopniu zmniejszały przerost lewej komory. Leczenie skojarzone aliskirenem i losartanem w porównaniu z monoterapią losartanem charakteryzowało się nieistotnie statystycznie wyraźniejszym zmniejszeniem wskaźnika masy lewej komory.2
Do badania Aliskiren in the Evaluation of Proteinuria in Diabetes (AVOID) włączono 599 chorych z nadciśnieniem tętniczym, cukrzycą typu 2 i cukrzycową chorobą nerek. Wyniki opublikowane w „New England Journal of Medicine” w czerwcu 2008 r. wskazują, że dodanie inhibitora reniny – aliskirenu – do dotychczas prowadzonego leczenia hipotensyjnego (zgodnie z protokołem chorzy otrzymywali antagonistę receptora angiotensyny II) u chorych z cukrzycą typu 2 i nadciśnieniem tętniczym zmniejsza białkomocz w porównaniu z placebo (–18% vs +2%). Autorzy tego badania podsumowują, że „aliskiren może mieć właściwości nefroprotekcyjne, niezależne od wpływu na wysokość ciśnienia tętniczego, u chorych na nadciśnienie tętnicze i cukrzycę typu 2 współistniejące z cukrzycową chorobą nerek, otrzymujących zalecane leczenie nefroprotekcyjne”.3
W grupie chorych z cukrzycą typu 2 i cukrzycową chorobą nerek również prowadzone jest badanie z użyciem aliskirenu – Aliskiren Trial in Type 2 Diabetes Using Cardio-Renal Disease Endpoints (ALTITUDE). Do badania włączonych zostanie około 8600 chorych na cukrzycę typu 2 z: makroalbuminurią i eGFR ≥30 ml/min/1,73m2 lub mikroalbuminurią i eGFR 30-60 ml/min/1,73m2, lub z rozpoznaną chorobą sercowo-naczyniową i eGFR 30-60 ml/min/1,73m2. W toku trwającej ok. 4 lat obserwacji oceniany będzie wpływ aliskirenu w porównaniu z placebo na częstość zdarzeń sercowo-naczyniowych i zdarzeń nerkowych.
Należy podkreślić, że wprowadzenie do stosowania klinicznego inhibitorów reniny stanowi nową możliwość leczenia nadciśnienia tętniczego. Staessen i wsp. postulują, że inhibitory reniny mogą być przydatne u chorych otrzymujących diuretyki, u których wzrasta aktywność reniny, u osób w młodym wieku ze wzmożoną aktywnością reninową, a także u chorych, u których inhibitory konwertazy angiotensyny powodują kaszel. Ich zdaniem mogą być też przydatne u osób z chorobami sercowo-naczyniowymi współistniejącymi z niewydolnością nerek. Wynika to z właściwości aliskirenu, który wydalany jest głównie z żółcią i w małym stopniu wchodzi w interakcje z innymi lekami.4
Luft i Weinberger zaznaczają, że w momencie wprowadzania do praktyki klinicznej antagonistów receptora angiotensyny II również zadawano pytania o wskazania do stosowania tej grupy leków. Wyniki dużych badań klinicznych dowiodły korzyści klinicznych i są podstawą formułowania stale poszerzanych wskazań do stosowania sartanów.5-7
Zalecenia ESH/ESC z 2007 r. zaznaczają miejsce inhibitorów reniny w terapii nadciśnienia tętniczego i podkreślają, że „aliskiren, nowy lek działający na poziomie układu reninowego, jest już dostępny w USA i wkrótce może być też dostępny w Europie [aliskiren został już zarejestrowany w krajach UE – przyp. aut.]. Lek ten skutecznie obniża ciśnienie krwi u chorych z nadciśnieniem, zarówno w monoterapii, jak i w skojarzeniu z diuretykiem tiazydowym, a w badaniach przedklinicznych zmniejszał białkomocz. Sugeruje się, że renina może powodować efekty niezwiązane z klasyczną kaskadą renina-angiotensyna i być czynnikiem rokowniczym niezależnym od wytwarzania angiotensyny II. Nie ma jeszcze danych, które by ostatecznie to potwierdzały, ani danych na temat korzystnych efektów sercowo-naczyniowych zahamowania reniny”.7
Piśmiennictwo
1. Oparil S, Yarows SA, Patel S, et al. Efficacy and safety of combined use of aliskiren and valsartan in patients with hypertension: a randomised, double-blind trial. Lancet 2007;370:221-229.
2. Solomon SD, Appelbaum E, Manning WJ, et al. Effect of the direct Renin inhibitor aliskiren, the Angiotensin receptor blocker losartan, or both on left ventricular mass in patients with hypertension and left ventricular hypertrophy. Circulation 2009;119:530-537.
3. Parving HH, Persson F, Lewis JB, et al. Aliskiren combined with losartan in type 2 diabetes and nephropathy. N Engl J Med 2008; 358:2433-2446.
4. Staessen JA, Li Y, Richart T. Oral renin inhibitors. Lancet 2006;368:1449-1456.
5. 2003 European Society of Hypertension-European Society of Cardiology guidelines for the management of arterial hypertension. J Hypertens 2003;21:1011-1053.
6. Luft FC, Weinberger MH. Antihypertensive therapy with aliskiren. Kidney Int 2007.
7. Mancia G, De Backer G, Dominiczak A, et al. 2007 Guidelines for the Management of Arterial Hypertension: The Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). J Hypertens 2007;25:1105-1187.