Program edukacyjny: hipertensjologia
Blokada receptora reninowego: lepsza strategia ochrony nerek niż hamowanie układu renina-angiotensyna-aldosteron?
Dominik N. Müller, PhD
Friedrich C. Luft, MD
W SKRÓCIE
Prorenina i renina wiążą się ze znajdującym się na powierzchni, lecz działającym wewnątrzkomórkowo, 350-aminokwasowym białkiem zwanym receptorem (pro)reninowym. Gdy N-koniec receptora wiąże się z proreniną lub reniną, uaktywnia się wewnątrzkomórkowy szlak sygnałowy, w którym pośredniczą regulowane zewnątrzkomórkowo kinazy, a wynikiem aktywacji może być produkcja inhibitora aktywatora plazminogenu i TGF-β. Badaczom udało się opracować nowatorski peptyd maskujący – peptyd regionu chwytnego (HRP – handle-region peptide) – który uniemożliwia wiązanie proreniny z receptorem. W badaniu HRP skutecznie hamował nefropatię cukrzycową u szczurów oraz u myszy pozbawionych receptorów angiotensynowych, a także zapobiegał włóknieniu mięśnia sercowego u szczurów z pierwotnym nadciśnieniem tętniczym. Ten sam zespół stworzył szczep transgenicznych szczurów, u których HRP wywołuje silną ekspresję receptorów (pro)reninowych i rozwój stwardnienia kłębuszków nerkowych. Niestety, grupy badawcze z Berlina, Paryża i Rotterdamu nie były w stanie potwierdzić tych wyników, a dokładne znaczenie receptorów (pro)reninowych nie jest znane.
Wstęp
Układ renina-angiotensyna okazał się najważniejszym systemem regulującym ciśnienie krwi i pracę układu krwionośnego, zwłaszcza z terapeutycznego punktu widzenia. Stworzenie najpierw inhibitorów konwertazy angiotensyny (ACE), a potem antagonistów receptora dla angiotensyny II (sartanów) i wreszcie bezpośrednich inhibitorów reniny pozwoliło lekarzom zgromadzić arsenał leków skutecznie walczących z reniną i jej jedynym znanym substratem – produktem przemiany angiotensynogenu. Niedawno pojawił się nowy możliwy cel dla farmakoterapii. Jego przedstawienie wymaga jednak nakreślenia tła historycznego.
W 1975 r. Day i wsp.1 opublikowali ciekawą pracę na temat „dużej” reniny – nieaktywnej i cięższej postaci hormonu, którą znaleźli u trzech pacjentów z nadciśnieniem i nefropatią. „Dużą” reninę (obecnie nazywaną proreniną) wykryto także w płynie owodniowym i w guzach Wilmsa. Proreninę może uaktywniać niskie pH. Autorzy sądzili, że to prorenina może być u niektórych pacjentów przyczyną nadciśnienia tzw. niskoreninowego.
Niedługo potem Luetscher i wsp.2,3 odkryli, że proreninę mogą też aktywować niskie temperatury. W dwóch niezwykle ciekawych, ale niedocenionych pracach autorzy opisali, że u chorych z cukrzycą stężenia krążącej we krwi proreniny są znacznie podwyższone, mimo niskiej aktywności osoczowej reniny (PRA). To stężenie proreniny, a nie aktywność osoczowa reniny, bezpośrednio korelowało z występowaniem powikłań cukrzycowych.3 Véniant i wsp.4 opisali spektakularne badanie, w którym stworzyli transgenicznego szczura wytwarzającego duże ilości proreniny. U zwierzęcia doszło do ciężkiej waskulopatii, pozornie bez zwyżki ciśnienia, choć nie przeprowadzono pomiarów telemetrycznych.
Dziś wiemy, że renina – proteaza aspartylowa – ma masę cząsteczkową ok. 40 kD.5 Jej cząsteczka składa się dwóch jednakowych płatów, a miejsce aktywne enzymatycznie znajduje się w szczelinie między nimi. Szczelina jest ukryta pod 43-aminokwasowym N-końcowym propeptydem o masie cząsteczkowej ok. 7 kD, który działa jak bramkarz i utrudnia dostęp angiotensynogenu. Prorenina jest syntetyzowana w postaci preprohormonu zawierającego peptyd sygnalny kierujący białko do retikulum endoplazmatycznego, a w końcu do miejsca syntezy na zewnątrz komórki. Prorenina może być uaktywniana proteolitycznie poprzez odcięcie propeptydu, które zachodzi w aparacie przykłębuszkowym, gdzie uwalniana jest aktywna renina. Do aktywacji może też dojść na drodze nieproteolitycznej – w wyniku działania kwasu lub mrożenia. Proreninę mogą też wytwarzać komórki poza nerką, np. oka, jądra, jajnika, gruczołów ślinowych, nadnerczy, hormonalnie czynnych guzów wytwarzających reninę oraz, co ciekawe, komórki tuczne.
Receptor (pro)reninowy
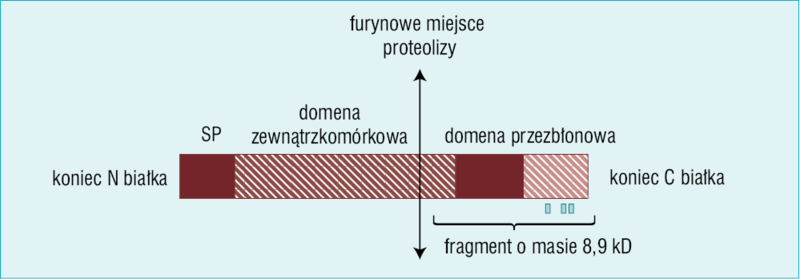
Rycina 1. Model receptora (pro)reninowego. W cząsteczce znajduje się furynowe miejsce proteolizy. Na rysunku zaznaczono przenikające przez błonę białko wakuolarnej ATPazy protonowej o masie 8,9 kD
Renina i prorenina mogą wiązać się z komórką w różnych miejscach, w tym poprzez receptor mannozo-6-fosforanowy oraz receptor dla insulinopodobnego czynnika wzrostu II.5 Nguyen i wsp.6 sklonowali receptor o wysokim powinowactwie do reniny i proreniny, który okazał się 350-aminokwasowym białkiem z pojedynczą domeną przezbłonową (ryc. 1).
Część tego białka wykazuje ścisłą (95%) homologię z poznanym wcześniej naczyniowym białkiem ATP6ap2 (związanym z częścią błonową ATPazy protonowej). Część ATP6ap2, początkowo określaną mianem M8-9, zidentyfikowano najpierw jako czynnik wyodrębniany wraz z fragmentem V0 ATPazy wakuolarnej w chromochłonnych błonach wołowych nadnerczy. Samo białko pełni funkcję adaptacyjną dla ATPazy protonowej. Nguyen i wsp.6 w swojej pracy dokonali jeszcze dwóch innych istotnych obserwacji. Gdy prorenina wiązała się z receptorem, cząsteczka ulegała aktywacji nieproteolitycznej, a zatem aktywowana prorenina (czyli aktywna renina) poprzez wiązanie mogła wytwarzać w tkankach angiotensynę II. Ponadto przypuszczalny receptor (pro)reninowy pośredniczył w aktywacji kinazy regulowanej przez sygnały wewnątrz- i zewnątrzkomówkowe kinazy (ERK1/2 – extracellular-related kinaze), która jest przedmiotem tego artykułu. Aktywacja ta mogła z kolei uruchamiać łańcuch kolejnych sygnałów i prowadzić do wytwarzania inhibitora-1 aktywatora plazminogenu (PAI-1) oraz transformującego czynnika wzrostu α (TGF-α). Co więcej, proces ten mógł zachodzić w sposób całkowicie niezależny od tworzenia angiotensyny II.7,8
Blokada receptorów reninowych
Suzuki i wsp.9 otrzymali przeciwciała skierowane przeciwko różnym fragmentom propeptydu („bramkarza”) i wysunęli hipotezę, że prorenina ma regiony bramkowe i chwytne, gdzie dochodzi do aktywacji nieproteolitycznej. Region chwytny (podobnie jak prawdopodobnie jeszcze inny region cząsteczki reniny) reaguje z receptorem (pro)reninowym. Po nich Ichihara i wsp.10 opublikowali pierwszy ze znakomitej serii artykułów poświęconych peptydowi pułapkowemu o sekwencji aminokwasowej RILLKKMPSV, który nazwali właśnie peptydem regionu chwytnego (HRP – handle-region peptide). Autorzy podawali ten peptyd podskórnie szczurom z cukrzycą wywołaną streptozotocyną. Badali osocze i nerki zwierząt po 8, 16 i 24 tygodniach od podania antybiotyku. Nerki szczurów z cukrzycą zawierały więcej angiotensyny I i II mimo braku zmian stężenia reniny, ACE i angiotensynogenu. Podawanie HRP całkowicie zahamowało rozwój nefropatii cukrzycowej mimo braku wpływu na hiperglikemię.
W kolejnej pracy badacze podawali HRP szczurom z idiopatycznym nadciśnieniem i skłonnością do udarów (Spontaneously Hypertensive Stroke Prone, SHR-SP) i dzięki leczeniu nie dochodziło do zwłóknienia mięśnia sercowego.11 Jeszcze bardziej spektakularne okazały się wyniki badań, w których podawanie HRP całkowicie uwalniało myszy z cukrzycą wywołaną delecją genu kodującego receptor AT1A od nefropatii cukrzycowej, konkretnie od białkomoczu i stwardnienia kłębuszków nerkowych.12 Wykazano w tym badaniu, że HRP działa nawet przy braku receptorów AT1A. W kolejnym badaniu na szczurach SHR-SP badacze wykazali, że ciągły podskórny wlew z HRP całkowicie hamował zarówno nieproteolityczną aktywację proreniny w tkankach, jak i pobudzenie tkankowego układu renina-angiotensyna, pozostając bez wpływu na stężenie krążącej reniny czy ciśnienie tętnicze.13 Leczenie znacząco łagodziło rozwój i postęp białkomoczu oraz twardnienia kłębuszków nerkowych.
Ci sami badacze stworzyli następnie transgeniczne szczury z nadekspresją ludzkiego receptora (pro)reninowego.14 Zwierzęta miały prawidłowe ciśnienie krwi oraz stężenie glukozy i angiotensyny II w osoczu, mimo to w miarę starzenia się rozwijał się u nich białkomocz, a w wieku 28 tygodni widoczne było stwardnienie kłębuszków nerkowych. Wlew z HRP w sposób znaczący hamował rozwój stwardnienia kłębuszków, nasilenie białkomoczu, aktywację kinazy białkowej aktywowanej przez mitogeny (mitogen-activated protein kinase – MAPK) oraz ekspresję TGF-β w nerkach. Podobnego działania nie obserwowano podczas leczenia inhibitorami ACE, mimo że leki te obniżały stężenie angiotensyny II. Rekombinowana szczurza prorenina pobudzała ponadto aktywację MAPK w hodowlach ludzkich komórek wykazujących ekspresję receptora (pro)reninowego, ale nie były one zdolne do aktywacji proreniny szczurzej. W podsumowaniu autorzy pracy stwierdzili, że w sposób jasny i klarowny dowiedli in vivo, że ludzkie receptory (pro)reninowe wywołują niezależną od angiotensyny II aktywację MAPK oraz że nasilona ekspresja TGF-β wywołuje stwardnienie kłębuszków nerkowych.
Inne ciekawe odkrycie z badań grupy Takahashiego15 dotyczy możliwości cofania się nefropatii cukrzycowej. Badacze 17-tygodniowym szczurom po heminefrektomii i z cukrzycą wywołaną podawaniem streptozotocyny, z podwyższonym stężeniem białka w moczu i znacznym stwardnieniem kłębuszków nerkowych podawali przez 12 tygodni HRP, inhibitor ACE lub placebo. Terapia HRP hamowała postęp cukrzycowych zmian w nerkach, podczas gdy inhibitor ACE jedynie w niewielkim stopniu spowalniał ich progresję. W nerkach szczurów z cukrzycą, którym podawano placebo, ekspresja aktywowanej proreniny była podwyższona, ale u gryzoni leczonych HRP – znacząco obniżona. Inhibitor ACE nie miał wpływu na ekspresję proreniny. Na podstawie tych wyników autorzy wysunęli wniosek, że blokada receptorów (pro)reninowych u szczurów z cukrzycą nie tylko hamuje postęp nefropatii, ale także odwraca stwardnienie kłębuszków nerkowych.
Ta sama grupa badaczy opublikowała wyniki prac na komórkach COS-7 transfekowanych receptorem (pro)reninowym.16 Wynikało z nich, że współczynnik Kd (równowagowego rozdziału stężeń) wynosi 0,89 i 1,8 nM. Naukowcy opisali też aktywację ludzkiej i szczurzej proreniny przez te komórki, którą można było zablokować za pomocą HRP. W jeszcze innym badaniu zespół starał się ustalić mechanizm, w którym ludzkie receptory (pro)reninowe aktywują MAPK w ludzkich komórkach mięśni gładkich naczyń. Zastosowano rekombinowaną ludzką proreninę, która nasilała ich proliferację i aktywowała ERK1/2 zależnie od czasu i dawki, nie wpływając przy tym na aktywację p38 ani c-Jun NH(2)-końcową kinazę.17 Stężenie aktywowanej ERK obniżono do poziomu kontrolnego za pomocą genisteiny – inhibitora kinazy tyrozynowej. Podobnie U0126 – inhibitor kinazy MAP kinazy (MEK) znacząco obniżał poziom aktywowanej ERK (do wyjściowego), podczas gdy na poziom ten nie wpływało podawanie inhibitorów ACE ani sartanów. Wyłączenie receptora (pro)reninowego przy użyciu małych blokujących odcinków kwasu rybonukleinowego(siRNA – small interfering RNA) znamiennie hamowało aktywację ERK wywołaną proreniną. Zdaniem autorów pracy zależne od receptora (pro)reninowego przenoszenie sygnału ERK może stać się nowym celem dla terapii zapobiegających powikłaniom naczyniowym.
Nie do przecenienia jest znaczenie odkryć zespołu Ichihary. Pozwalają one wyjaśnić obserwacje poczynione w laboratorium Luetschera1-3 i – przynajmniej częściowo – spektakularne rezultaty badania opublikowanego przez Vénianta i wsp.4 Co istotniejsze, ujawniają niesamowite możliwości terapeutyczne. Cukrzyca staje się najczęstszą przyczyną niewydolności nerek na świecie, i to mimo stosowania inhibitorów ACE, sartanów czy bezpośrednich inhibitorów reniny. Opracowanie możliwego do wykorzystania w praktyce inhibitora receptora (pro)reninowego (np. HRP) mogłoby stanowić przełom terapeutyczny. Tworzenie takich inhibitorów powinno być priorytetem dla każdej firmy farmaceutycznej. Istnieją jednak ku temu przeszkody, niemałe znaczenie ma tu to, że wyniki badań nie zostały potwierdzone przez niezależne laboratoria.
Feldt i wsp.18 porównali rezultaty działania aliskirenu (bezpośredniego inhibitora reniny) z HRP u podwójnie transgenicznych szczurów z nadekspresją zarówno genu dla ludzkiej reniny, jak i genu dla angiotensynogenu. Po 7 tygodniach żyły wszystkie gryzonie leczone aliskirenem, podczas gdy w grupie, której podawano HRP, śmiertelność wynosiła 58%, a w grupie kontrolnej – 40%. W przeciwieństwie do HRP aliskiren obniżał ciśnienie krwi i zmniejszał albuminurię, stężenie nystatyny C i lipokaliny neutrofilowej związanej z żelatynazą (wskaźnik uszkodzenia kanalików nerkowych) do poziomów obserwowanych u kontrolnych, nietransgenicznych szczurów. Ludzka renina i prorenina in vitro pobudzały wkomórkach mięśni gładkich naczyń fosforylację ERK1/2, niezależnie od angiotensyny II.
Preinkubacja z HRP lub aliskirenem nie zapobiegała fosforylacji ERK1/2 indukowanej zarówno przez reninę, jak i proreninę, podczas gdy inhibitor kinazy MAP (MEK1/2) PD98059 zapobiegał obydwu.