



Spis treści
Choroba Creutzfeldta-Jakoba jest jednostką chorobową o bardzo różnorodnej ekspresji klinicznej, sprawiającą duże trudności diagnostyczne. Lekarze w diagnostyce różnicowej swoich pacjentów powinni ją uwzględniać i sięgać po inne niż standardowe metody rozpoznawania tej choroby.
Wprowadzenie
Choroba Creutzfeldta-Jakoba (CJD – Creutzfeldt-Jakob disease) jest śmiertelną chorobą neurodegeneracyjną mózgu. Rozwija się w wyniku zadziałania czynnika nazywanego prionem (PrP – proteinaceous infectious
particle). Prion to prawidłowe komórkowe białko PrPc po patologicznej konwersji, polegającej na zmianie jego struktury przestrzennej do PrPSc. Wyróżniamy cztery podstawowe postacie CJD zależne od etiologii:
- sporadyczna (sporadic; sCJD) – rozwija się w wyniku spontanicznej zmiany konformacji białka prionowego
- rodzinna, (familial; fCJD) − powstaje w wyniku mutacji genu PRNP kodującego białko prionu
- jatrogenna (iatrogenic; iCJD) – związana z działaniami medycznymi (np. podanie hormonu wzrostu uzyskanego z przysadki pacjenta zakażonego formą sporadyczną choroby)
- wariant (variant; vCJD) − wywołana przeniesieniem encefalopatii gąbczastej bydła na człowieka przez spożycie zakażonych produktów pochodzenia zwierzęcego.
Najczęstszą postacią jest sCJD, stanowiąca około 85-95% wszystkich przypadków choroby Creutzfeldta-Jakoba. 1 Choroba sprawia duże trudności diagnostyczne, co wynika z faktu, że jest jednostką bardzo niejednorodną. Poszczególne przypadki różnią się objawami klinicznymi, przebiegiem i wynikami badań dodatkowych. Najczęstsze objawy to: szybko postępujące otępienie, zaburzenia móżdżkowe, zaburzenia wzrokowe, objawy uszkodzenia układu piramidowego lub pozapiramidowego, a także mioklonie, które często pojawiają się jednak dopiero w późnej fazie choroby. Okres inkubacji wynosi zazwyczaj od kilku do kilkunastu lat, a zgon następuje najczęściej od kilku do kilkunastu miesięcy od wystąpienia pierwszych objawów klinicznych. Najczęstszą przyczyną zgonu jest odoskrzelowe zapalenie płuc. Najistotniejszymi badaniami wykorzystywanymi w diagnostyce CJD są: rezonans magnetyczny mózgu, badanie elektroencefalograficzne (EEG) oraz badanie płynu mózgowo-rdzeniowego (PMR). Kryteria diagnostyczne sporadycznej postaci choroby Creutzfeldta-Jakoba przedstawiono w tabeli 1.
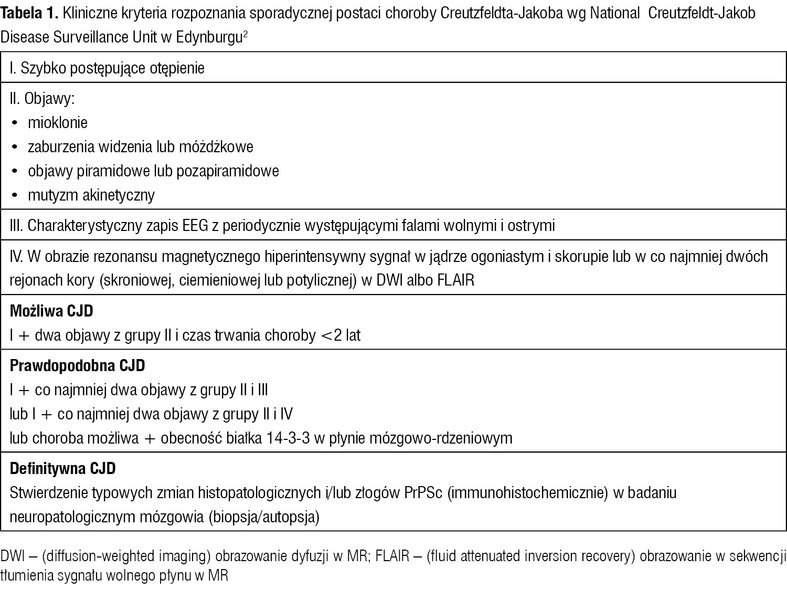
Tabela 1. Kliniczne kryteria rozpoznania sporadycznej postaci choroby Creutzfeldta-Jakoba wg National Creutzfeldt-Jakob Disease Surveillance Unit w Edynburgu2
Dodatni wynik testu pozwala na rozpoznanie prawdopodobnej CJD, definitywnie chorobę nadal rozpoznajemy tylko na podstawie badania neuropatologicznego.
Opis przypadku 1
Pacjentka 60-letnia została przyjęta na oddział neurologii w związku z wystąpieniem szybko postępujących zaburzeń funkcji poznawczych. Chorą przeniesiono z oddziału psychiatrii, gdzie przebywała z powodu labilności emocjonalnej, halucynacji, zachowań agresywnych. Objawy narastały od około czterech miesięcy.
W badaniu neurologicznym w dniu przyjęcia na oddział chora była podsypiająca, nie odpowiadała na pytania, nie spełniała poleceń, w reakcji na ból okresowo otwierała oczy, broniła się symetrycznie kończynami górnymi i dolnymi; napięcie mięśniowe było wzmożone plastycznie w kończynach górnych i dolnych. Pacjentka leżąca i zacewnikowana ze względu na dysfagię była karmiona przez sondę dożołądkową.
W badaniu MR głowy uwidoczniono odcinkowe podwyższenie intensywności sygnału w sekwencji obrazowania dyfuzji (DWI – diffusion-weighted imaging) i obrazowania tłumienia sygnału wolnego płynu (FLAIR – fluid attenuated inversion recovery) w korze płatów potylicznych i ciemieniowych (bez restrykcji dyfuzji) (ryc. 1A) oraz dyskretne, symetryczne podwyższenie sygnału w sekwencjach T2 i FLAIR w jądrach podkorowych, zwłaszcza w części grzbietowej obu wzgórz (ryc. 1B i C). W EEG zarejestrowano periodyczne kompleksy fali ostrej z falą wolną, z przewagą po stronie prawej. W badaniu płynu mózgowo-rdzeniowego stwierdzono obecność białka 14-3-3. Badanie ogólne PMR było prawidłowe, nie stwierdzono prążków oligoklonalnych, synteza wewnątrztekalna była ujemna. W surowicy i PMR nie stwierdzono przeciwciał przeciwko Borrelia burgdorferi. Ujemne były również wyniki badań na obecność autoprzeciwciał paranowotworowych [Hu, Yu, Ri, PNMA2(Ma2/Ta), CV2, amfifizyna], przeciwciał anty-NMDA, przeciwciał przeciwko peroksydazie tarczycowej i przeciwko tyreoglobulinie.
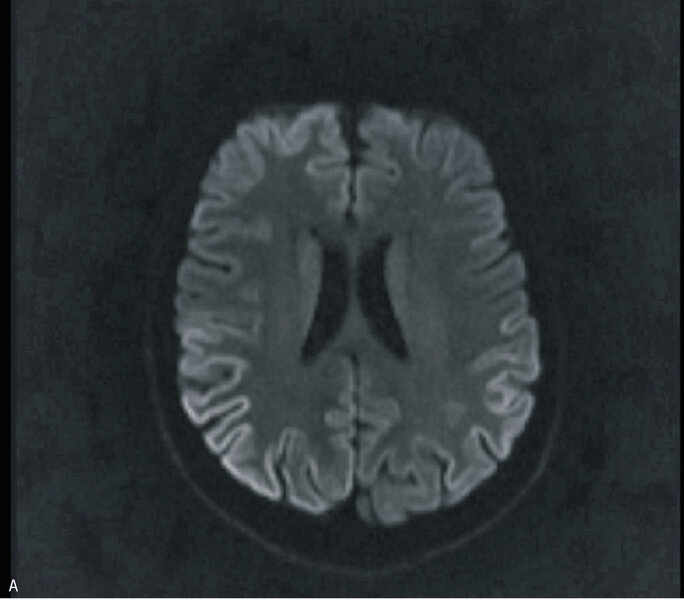
Rycina 1. Badanie MR mózgu – przypadek kliniczny 1. Sekwencja DWI (A). Odcinkowe podwyższenie intensywności sygnału w korze płatów potylicznych i ciemieniowych (bez restrykcji dyfuzji). Obraz T2-zależny (B). Sekwencja FLAIR. Symetryczne podwyższenie sygnału w jądrach podkorowych. Sekwencja T2 (C). Symetryczne podwyższenie sygnału w jądrach podkorowych
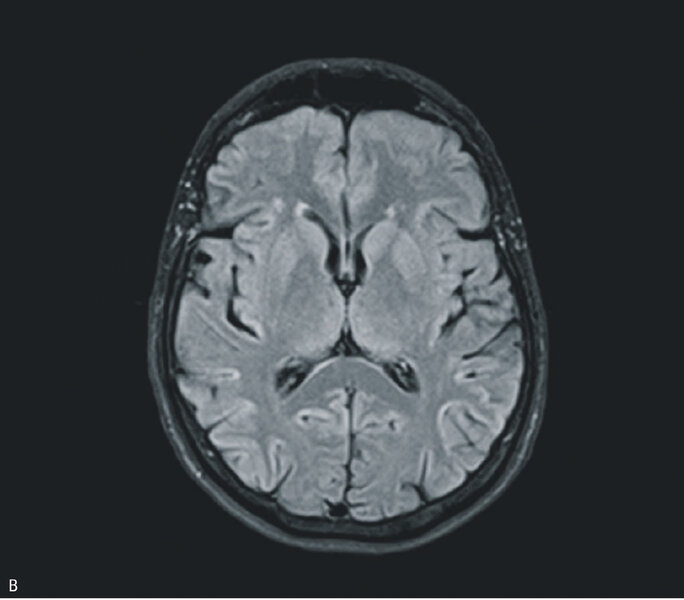
Rycina 1. Badanie MR mózgu – przypadek kliniczny 1. Sekwencja DWI (A). Odcinkowe podwyższenie intensywności sygnału w korze płatów potylicznych i ciemieniowych (bez restrykcji dyfuzji). Obraz T2-zależny (B). Sekwencja FLAIR. Symetryczne podwyższenie sygnału w jądrach podkorowych. Sekwencja T2 (C). Symetryczne podwyższenie sygnału w jądrach podkorowych
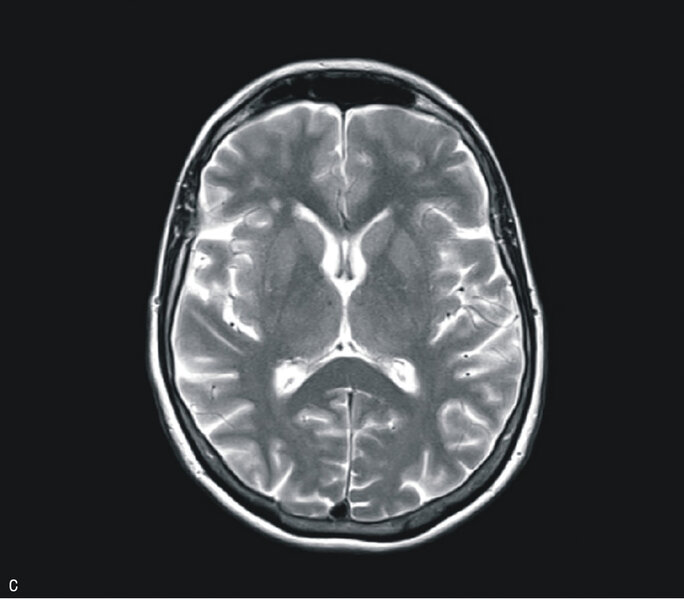
Rycina 1. Badanie MR mózgu – przypadek kliniczny 1. Sekwencja DWI (A). Odcinkowe podwyższenie intensywności sygnału w korze płatów potylicznych i ciemieniowych (bez restrykcji dyfuzji). Obraz T2-zależny (B). Sekwencja FLAIR. Symetryczne podwyższenie sygnału w jądrach podkorowych. Sekwencja T2 (C). Symetryczne podwyższenie sygnału w jądrach podkorowych
Obraz kliniczny oraz wyniki badań dodatkowych przemawiały za rozpoznaniem choroby Creutzfeldta-Jakoba – chora spełniła kryteria prawdopodobnej CJD.
Opis przypadku 2
Pacjentka 62-letnia została przeniesiona z oddziału chorób wewnętrznych na oddział neurologii z powodu szybko postępującego zespołu móżdżkowego oraz zaburzeń funkcji poznawczych. Objawy narastały od około pięciu miesięcy. Początkowo chora miała zaburzenia równowagi, upadała, w chwili przyjęcia nie była w stanie chodzić samodzielnie. Syn pacjentki zgłaszał, że kobieta okresowo nie poznaje członków rodziny, jest niezorientowana w kwestii podstawowych informacji.
W badaniu neurologicznym w dniu przyjęcia na oddział chora była przytomna, prawidłowo zorientowana auto- i allopsychicznie, stwierdzono u niej oczopląs poziomy przy patrzeniu na boki, ataksję kończyn górnych i dolnych, ataksję tułowia i chodu. Przy próbie pionizacji kobieta upadała do tyłu, okresowo widoczne były mioklonie tułowia i kończyn.
W MR głowy stwierdzono cechy obustronnej restrykcji dyfuzji w obrębie głów i trzonów jąder ogoniastych oraz w mniejszym stopniu w zakresie skorupy, z dyskretnym poszerzeniem obrysów głowy prawego jądra ogoniastego; zmiany były obustronne i symetryczne (ryc. 2A, B i C). Zapis EEG nie był charakterystyczny dla CJD. Wynik był nieprawidłowy z rejestracją synchronicznych fal Δ w odprowadzeniach czołowych (FRIDA) na tle średniego stopnia uogólnionego zwolnienia czynności podstawowej. Pacjentka była badana neuropsychologicznie. Całość badania ujawniła zaburzenia funkcji poznawczych. Poziom nasilenia objawów wskazywał na otępienie lekkiego stopnia. Wyniki badania ogólnego płynu mózgowo-rdzeniowego były prawidłowe, nie stwierdzono obecności prążków oligoklonalnych, synteza wewnątrztekalna była ujemna. Wyniki badań w kierunku chorób zakaźnych (kiła, HIV) były ujemne. Badanie markerów nowotworowych (Ca 125, Ca 19-9, CEA) oraz badanie w kierunku obecności autoprzeciwciał paranowotworowych [Hu, Yu, Ri, PNMA2(Ma2/Ta), CV2, amfifizyna] były ujemne. Stężenia ceruloplazminy, witaminy B12 i kwasu foliowego były w normie.
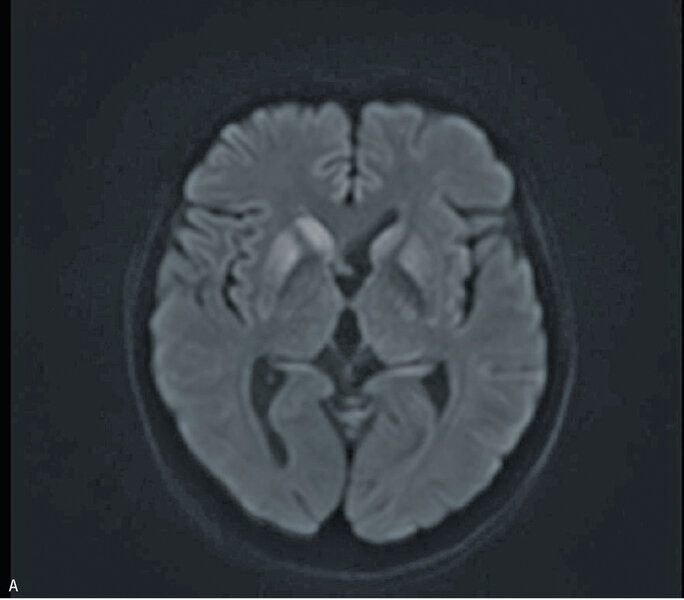
Rycina 2. Badanie MR mózgu – przypadek kliniczny 2. Sekwencja DWI (A). Cechy obustronnej restrykcji dyfuzji w obrębie głów i trzonów jąder ogoniastych oraz w zakresie skorupy, z dyskretnym poszerzeniem obrysów głowy prawego jądra ogoniastego. Sekwencja ADC (B). Obraz T2-zależny (C). Sekwencja FLAIR
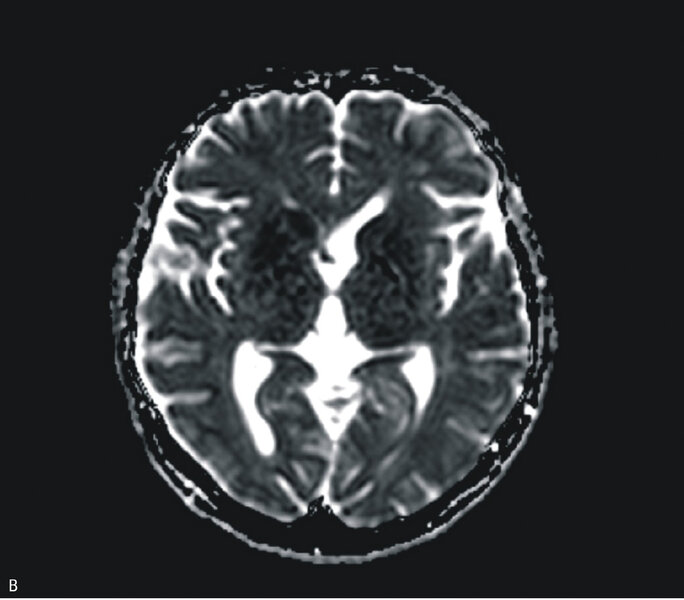
Rycina 2. Badanie MR mózgu – przypadek kliniczny 2. Sekwencja DWI (A). Cechy obustronnej restrykcji dyfuzji w obrębie głów i trzonów jąder ogoniastych oraz w zakresie skorupy, z dyskretnym poszerzeniem obrysów głowy prawego jądra ogoniastego. Sekwencja ADC (B). Obraz T2-zależny (C). Sekwencja FLAIR
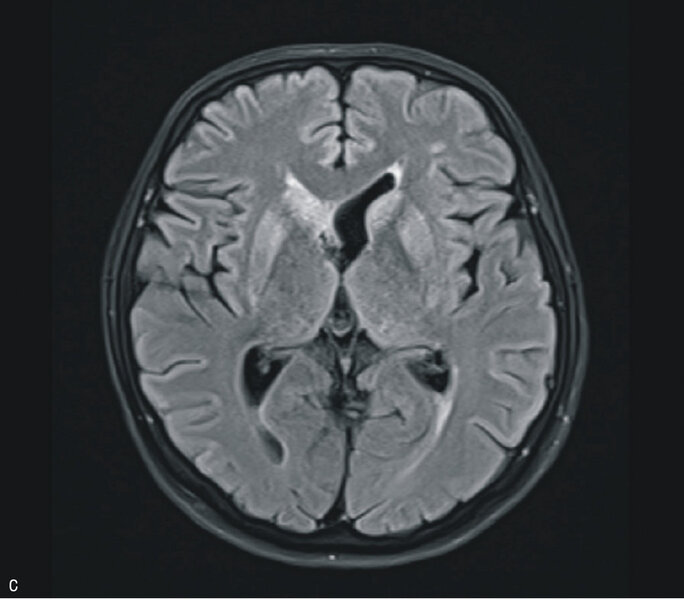
Rycina 2. Badanie MR mózgu – przypadek kliniczny 2. Sekwencja DWI (A). Cechy obustronnej restrykcji dyfuzji w obrębie głów i trzonów jąder ogoniastych oraz w zakresie skorupy, z dyskretnym poszerzeniem obrysów głowy prawego jądra ogoniastego. Sekwencja ADC (B). Obraz T2-zależny (C). Sekwencja FLAIR
U chorej wysunięto podejrzenie CJD, ponieważ spełniła kryteria możliwej choroby. Wysłano próbkę płynu mózgowo-rdzeniowego do laboratorium w Edynburgu na badanie w kierunku obecności białka prionowego (RT-QuIC), którego wynik okazał się dodatni; w PMR stwierdzono również obecność białka 14-3-3.
Opis przypadku 3
Pacjentka 57-letnia została przyjęta na oddział neurologii z powodu osłabienia siły mięśniowej oraz ruchów mimowolnych kończyn lewych, zaburzeń równowagi i chodu. Powyższe objawy narastały od około 2 miesięcy.
W badaniu neurologicznym w dniu przyjęcia na oddział chora była przytomna, zorientowana prawidłowo auto- i allopsychicznie. Stwierdzono u niej dyzartrię, niskiego stopnia proksymalny niedowład kończyn lewych i ich ruchy mimowolne o charakterze dystonicznym, przeczulicę na kończynach i tułowiu po stronie lewej, ataksję tułowia, kończyny górnej lewej oraz obu kończyn dolnych, chód ataktyczny w asekuracji dwóch osób. Podczas hospitalizacji obserwowano szybką progresję objawów, szczególnie bardzo szybki postęp zaburzeń funkcji poznawczych, dołączyły się również ataksja oraz dystonia w prawej kończynie górnej, dysfagia, mioklonie oraz uogólnione i częściowe napady padaczkowe.
Badanie MR głowy było trudne do oceny ze względu na artefakty, nie uwidoczniono jednak ewidentnych nieprawidłowości (ryc. 3A, B i C). W kilkukrotnie wykonanych badaniach EEG zarejestrowano ewolucję zmian w kierunku periodycznych, tłumionych przez senność kompleksów fali ostrej z falą wolną. W badaniu neuropsychologicznym stwierdzono zaburzenia funkcji poznawczych oraz ich dynamiczną progresję widoczną w ciągu zaledwie kilku dni. W płynie mózgowo-rdzeniowym wykryto obecność białka 14-3-3. Badanie ogólne PMR było prawidłowe, nie stwierdzono obecności prążków oligoklonalnych, synteza wewnątrztekalna była ujemna. W surowicy i PMR nie stwierdzono przeciwciał przeciwko Borrelia burgdorferi. Ujemne były również wyniki badań na obecność autoprzeciwciał paranowotworowych [Hu, Yu, Ri, PNMA2(Ma2/Ta), CV2, amfifizyna], przeciwciał anty-NMDA, przeciwciał antykardiolipinowych oraz przeciwciał przeciwko endomyzjum. Wyniki badań w kierunku zakażeń wirusami neurotropowymi (EBV, CMV, HSV, VZV, enterowirusy) były ujemne. Obraz kliniczny i wyniki badań dodatkowych przemawiały za rozpoznaniem sCJD – chora spełniła kryteria prawdopodobnej CJD. W celu weryfikacji rozpoznania wysłano próbkę płynu mózgowo-rdzeniowego na badanie w kierunku obecności białka prionowego (RT-QuIC) do ośrodka w Getyndze i Edynburgu – oba wyniki badań były dodatnie.
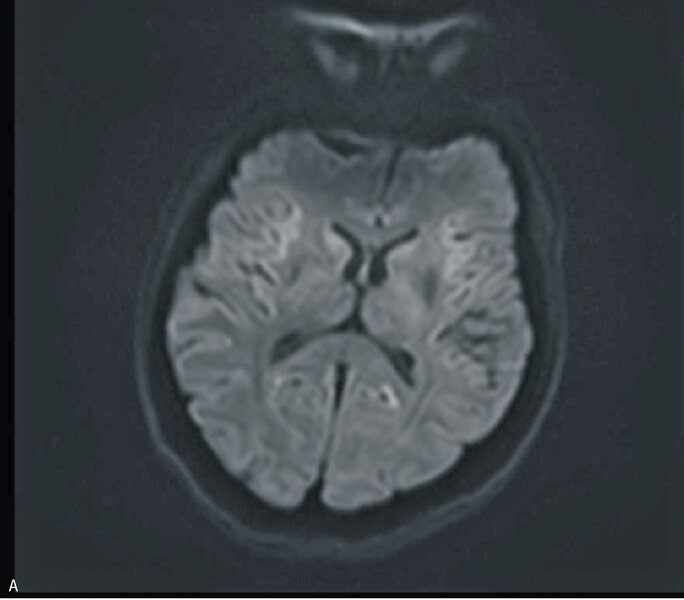
Rycina 3. Badanie MR mózgu – przypadek kliniczny 3. Sekwencja DWI (A). Sekwencja ADC (B). Obraz T2-zależny (C). Sekwencja FLAIR
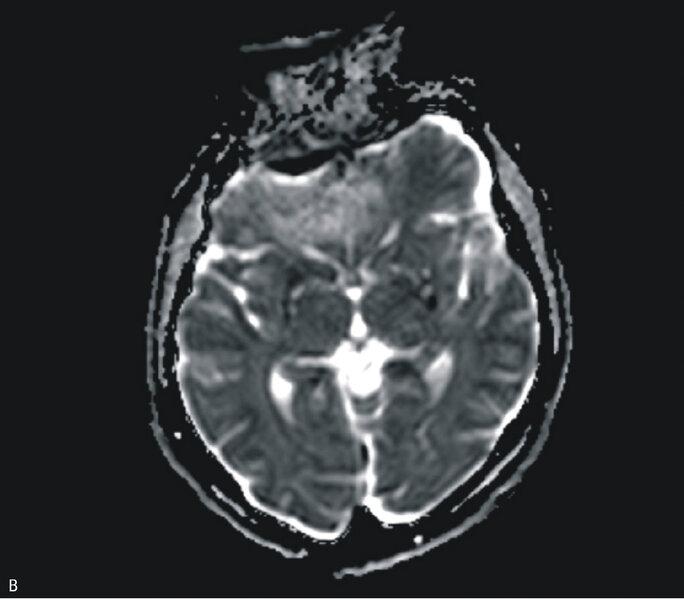
Rycina 3. Badanie MR mózgu – przypadek kliniczny 3. Sekwencja DWI (A). Sekwencja ADC (B). Obraz T2-zależny (C). Sekwencja FLAIR
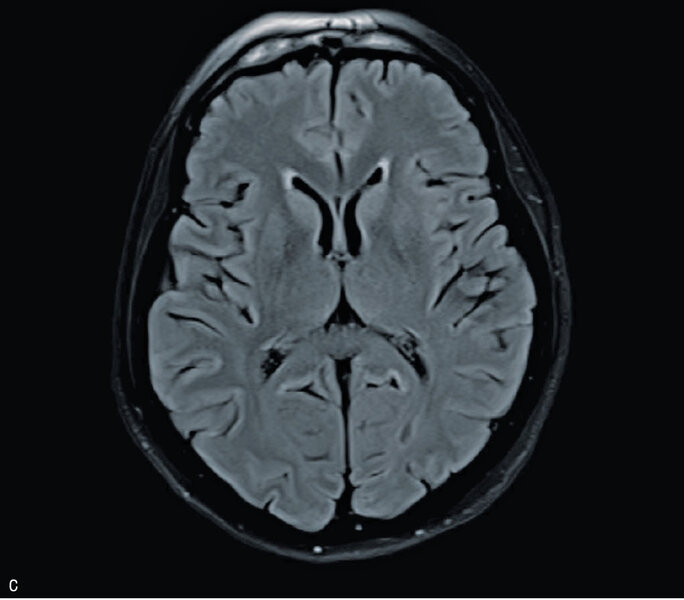
Rycina 3. Badanie MR mózgu – przypadek kliniczny 3. Sekwencja DWI (A). Sekwencja ADC (B). Obraz T2-zależny (C). Sekwencja FLAIR
Podsumowanie
Opis powyższych przypadków klinicznych potwierdza, że CJD jest chorobą o bardzo różnorodnej ekspresji klinicznej, sprawiającą duże trudności diagnostyczne. Zachęcamy lekarzy, aby w diagnostyce różnicowej swoich pacjentów uwzględniali tę jednostkę chorobową oraz sięgali po inne niż standardowe metody diagnostyczne, takie jak test RT-QuIC.
Abstract
Creutzfeldt-Jakob disease – Presentation of clinical cases
Creutzfeldt-Jakob disease (CJD) is a fatal neurodegenerative disorder. CJD is caused by a protein called a prion (proteinaceous infectious particle, PrP). Prion is a normal cellular prion protein (PrPC) converted into a disease-related isoform (PrPSc) due to misfolding. The most common form of the disease – sporadic CJD – accounts for about 90% of all recognized CJD cases. The disease is very difficult to diagnose due to heterogeneous clinical presentation. Individual cases are characterized by different clinical symptoms, clinical course and results of additional tests. This paper presents 3 clinical cases of patients hospitalized at the Neurology Department at a specialist hospital in Cracow, Poland. In 2 cases, the standard diagnostic procedure was extended to include RT-QuIC (Real-Time Quaking-Induced Conversion) analysis, which is not available in Poland, for the detection of prion protein in the cerebrospinal fluid. Compared to 14-3-3 protein detection, RT-QuIC test has a higher diagnostic value. The 14-3-3 protein is a neuronal protein which is released into the cerebrospinal fluid in response to neuronal damage, and therefore it can be found in a number of different neurological disorders. RT-QuIC test detects an abnormal prion protein which is a specific marker for CJD. The sensitivity of the test is estimated at 85% and its specificity – at 100%.
- 1. Heinemann U, Krasnianski A, Meissner B, et al. Creutzfeldt-Jakob disease in Germany: a prospective 12-year surveillance. Brain 2007;130:1350-9.
- 2. National Creutzfeldt-Jakob Disease Surveillance Diagnostic Criteria. National Creutzfeldt-Jakob Disease Surveillance 2010. http://www.cjd.ed.ac.uk.
- 3. McGuire L, Peden AD, Orru C, et al. Real Time Quaking-Induced Conversion Analysis of Cerebrospinal Fluid in Sporadic Creutzfeldt-Jakob Disease. American Neurological Association 2012;72:278-85.
- 4. Peden AH, McGuire LI, Appleford NE. Sensitive and specific detection of sporadic Creutzfeldt-Jakob disease brain prion protein using real-time quaking-induced conversion. J Gen Virol 2012;93:438-49.
- 5. Atarashi R, Sano K, Satoh K, Nishida N. Real-time quaking-induced conversion. A highly sensitive assay for prion detection. Prion 2011;5(3):150-3.
- 6. Atarashi R, Satoh K, Sano K, et al. Ultrasensitive human prion detection in cerebrospinal fluid by real-time quaking-induced conversion. Nat Med 2011;17(2):175-8.
Następny artykuł:
Zanik korowy tylny w świetle nowych kryteriów diagnostycznych

 Dodaj do ulubionych
Dodaj do ulubionych
