Pacjent w trakcie leczenia onkologicznego
Ostra białaczka szpikowa – diagnostyka i leczenie
dr hab. n. med. Łukasz Bołkun

dr hab. n. med. Łukasz Bołkun
- Patogeneza ostrej białaczki szpikowej i konieczne do jej potwierdzenia badania diagnostyczne
- Omówienie leczenia białaczki szpikowej z podziałem na chorych przed 60 rokiem życia i starszych
Definicja i patogeneza
Ostra białaczka szpikowa (AML – acute myleoid leukemia) jest najczęstszą ostrą białaczką występującą u dorosłych i stanowi ok. 80% wszystkich zachorowań. Zapadalność na AML wynosi ok. 3,7 przypadku na 100 000 mieszkańców na rok i zwiększa się znacząco wraz z wiekiem. U pacjentów po 65 r.ż. jest już kilkukrotnie wyższa i wynosi 10 zachorowań na 100 000 mieszkańców na rok, natomiast w przypadku pacjentów po 80 r.ż. ten wskaźnik przewyższa 25 przypadków na 100 000 osób na rok. Mężczyźni chorują nieznacznie częściej niż kobiety (odpowiednio 4,56 vs 3,0 na 100 000 na rok)1,2.
Ostra białaczka szpikowa jest nowotworem złośliwym wywodzącym się z linii białokrwinkowej, w której dochodzi do proliferacji i kumulacji w szpiku kostnym niedojrzałych, nieprawidłowych komórek blastycznych, pochodzących ze stransformowanej nowotworowo prekursorowej komórki mieloidalnej3,4. Nagromadzenie się komórek blastycznych, których cechą jest brak dojrzałości zarówno pod względem morfologicznym, jak i czynnościowym, prowadzi do wyparcia prawidłowej hematopoezy ze szpiku kostnego2.
Objawy kliniczne najczęściej występują 2-8 tygodni przed ustaleniem rozpoznania. Najważniejsze i najczęstsze są związane z obecnością komórek blastycznych w jamie szpikowej, gdzie ich niekontrolowana proliferacja skutkuje wyparciem prawidłowej hematopoezy. W efekcie występują symptomy wynikające z niewydolności prawidłowej hematopoezy, co objawia się: niedokrwistością (głównie pod postacią bólów i zawrotów głowy, pogorszenia stanu układu sercowo-naczyniowego, znacznego osłabienia), małopłytkowością (główne objawy to skaza małopłytkowa w postaci wybroczyn na skórze i błonach śluzowych, krwawień z nosa, dziąseł czy przedłużającego się u kobiet krwawienia miesiączkowego). W skrajnych przypadkach małopłytkowość może jednak prowadzić do ciężkich krwawień zagrażających życiu, w tym z przewodu pokarmowego czy do ośrodkowego układu nerwowego (OUN). Objawy ciężkiej skazy krwotocznej mogą również występować w przebiegu zespołu rozsianego wykrzepiania wewnątrznaczyniowego (DIC – disseminated intravascular coagulation), najczęściej stwierdzanego w podtypie ostrej białaczki szpikowej, tj. w ostrej białaczce promielocytarnej (APL – acute promyelocytic leukemia). Objawami związanymi z neutropenią w zależności od jej głębokości są głównie zakażenia grzybicze i bakteryjne w tkankach i narządach, niejednokrotnie przyczyniające się do zgonu pacjenta czy też uniemożliwiające włączenie leczenia. Mogą też wystąpić wyjątkowo niespecyficzne objawy ostrej białaczki, takie jak nagła utrata masy ciała czy bóle kostne1,4,5. W badaniu przedmiotowym znacznie rzadziej – w porównaniu z ostrą białaczką limfoblastyczną – stwierdza się powiększenie węzłów chłonnych, wątroby czy śledziony. Zajęcie OUN w postaci białaczkowego zapalenia opon mózgowo-rdzeniowych (meningitis leukemica) lub neurologicznych objawów ogniskowych obserwuje się najczęściej w podtypie AML ze zróżnicowaniem monocytarnym/monoblastycznym5.
Należy podkreślić, że nieleczona ostra białaczka szpikowa bardzo szybko prowadzi do niewydolności wielonarządowej, głównie spowodowanej ciężkimi infekcjami bakteryjnymi i grzybiczymi lub ciężkimi powikłaniami krwotocznymi, w tym krwawieniem do OUN.
Etiologia ostrej białaczki jest złożona i nie jest do końca znana. Do przyczyn mogących mieć wpływ na jej występowanie zalicza się wcześniejszą ekspozycję na czynniki środowiskowe, takie jak rozpuszczalniki (np. beznen), promieniowanie jonizujące, palenie papierosów, czy też wcześniejsze leczenie przy użyciu chemioterapeutyków, np. leków alkilujących czy inhibitorów topoizomerazy II. Jednocześnie stwierdza się zwiększone prawdopodobieństwo rozwoju AML w przebiegu licznych chorób o podłożu genetycznym, wśród których należy wyróżnić wrodzone defekty genetyczne, np. zespół Downa czy Blooma, oraz zespoły niewydolności szpiku, takie jak niedokrwistość Fanconiego czy deskeratoza wrodzona4, a także inne choroby układu krwiotwórczego mogące przekształcić się w AML, w tym przewlekłą białaczkę szpikową, czerwienicę prawdziwą czy mielofibrozę1,3,6.
W ciągu ostatnich kilkunastu lat dzięki możliwości zastosowania sekwencjonowania nowej generacji (NGS – next generation sequencing), nowoczesnych badań molekularnych dokonał się ogromny postęp w zrozumieniu biologii choroby i dużego zróżnicowania genetycznego AML. Dotąd opisano wiele czynników mogących predysponować do rozwoju ostrej białaczki. U podłoża rozwoju choroby leży kumulacja nabytych zaburzeń genetycznych powstałych w komórkach prekursorowych linii mieloidalnej, a przede wszystkim w genach zapewniających zachowanie równowagi między proliferacją a apoptozą komórek hematopoezy. Wśród najlepiej poznanych mutacji w genach odgrywających znamienną rolę w patogenezie zalicza się: mutacje w genach cząstek sygnałowych, np. FLT3, RAS, których stała aktywacja prowadzi do proliferacji komórek, mutacje w genach biorących udział w regulacji epigenetycznej (tj. geny biorące udział w regulacji modyfikacji DNA i regulacji stanu chromatyny, np. ASXL1, IDH1 i IDH2) czy też w genach fuzyjnych mieloidalnych komórek transkrypcyjnych [geny fuzyjne dla czynników transkrypcji, np. t(8;21), lub mutacje czynników transkrypcyjnych] prowadzące do zahamowania różnicowania komórek hematopoetycznych. Ich wykrycie przy rozpoznaniu staje się obecnie ważnym czynnikiem diagnostycznym, umożliwiającym dokładniejsze zróżnicowanie ostrej białaczki, ale również lepszy dobór leków w terapii celowanej AML7.
Badania diagnostyczne
Głównym badaniem, na podstawie którego można u pacjenta podejrzewać ostrą białaczkę, jest morfologia krwi obwodowej z ręcznym, mikroskopowym rozmazem krwi, w której przeważnie stwierdza się obecność niedokrwistości normocytarnej (wyjątek stanowią pacjenci z wcześniejszym zespołem mielodysplastycznym [MDS – mielodysplastic syndrome], u których niedokrwistość jest makrocytarna) i małopłytkowości w różnym stopniu nasilenia. Liczba białych krwinek jest różna, nawet u 30-40% pacjentów jest na granicy normy fizjologicznej, u pozostałych chorych stwierdza się leukopenię (szczególnie u pacjentów z wcześniejszym MDS) lub hiperleukocytozę, której wartość może przekraczać 200 lub 300 G/l4. Najcharakterystyczniejszą cechą takiej morfologii jest obecność w rozmazie krwi obwodowej młodych komórek, nazywanych komórkami blastycznymi, oraz resztkowych dojrzałych granulocytów. Brak from pośrednich lub ich nieznaczna liczba między komórkami blastycznymi a dojrzałymi tworzy tzw. przerwę białaczkową (hiatus leucaemicus).
Podstawowym badaniem wykonywanym w celu potwierdzenia ostrej białaczki szpikowej jest mielogram, badanie cytologiczne szpiku kostnego, barwionego za pomocą metody Maya-Grünwalda-Giemsy. W celu oceny szpiku zaleca się przeanalizowanie przynajmniej 500 komórek jądrzastych. Trepanobiopsja, która polega na pobraniu wycinka kostnego, nie jest zalecana standardowo i powinna być wykonana w przypadku braku możliwości uzyskania miarodajnego materiału (tzw. biopsja pusta). Według Światowej Organizacji Zdrowia w celu rozpoznania AML konieczne jest stwierdzenie co najmniej 20% komórek blastycznych w szpiku i/lub krwi obwodowej. Wyjątkiem są białaczki z powtarzalnymi aberracjami cytogentycznymi: t(8;21), inv(16), t(16;16) lub t(15;17)7.
W celu potwierdzenia ostrej białaczki szpikowej i dalszego różnicowania na poszczególne podtypy wykonuje się analizę cytometryczną komórek blastycznych, badania molekularne i cytogenetyczne. Obecnie w celu potwierdzenia zróżnicowania liniowego powszechnie wykorzystuje się metodę wielokolorowej cytometrii przepływowej, która umożliwia ocenę aberrantnych fenotypów białaczkowych, służących do zbadania i monitorowania tzw. choroby resztkowej, czyli stopnia eliminacji komórek blastycznych ze szpiku kostnego. Znany jest szeroki panel antygenów linii komórkowych, które umożliwiają określenie stopnia dojrzałości i zróżnicowania analizowanych komórek. Należą do nich markery prekursorowe, markery granulocytarne, monocytowe, megakariocytowe czy erytroidalne (tab. 1)6,8.
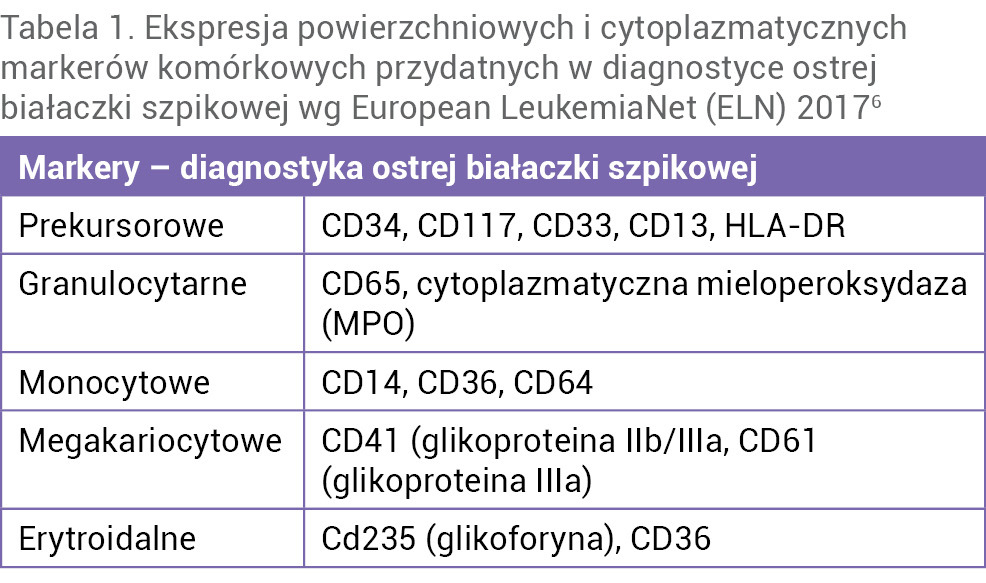
Tabela 1. Ekspresja powierzchniowych i cytoplazmatycznych markerów komórkowych przydatnych w diagnostyce ostrej białaczki szpikowej wg European LeukemiaNet (ELN) 20176
U każdego pacjenta należy wykonać klasyczne badanie cytogenetyczne metodą prążkową, która umożliwia ocenę kariotypu komórek białaczkowych, a ich aberracje mają zastosowanie prognostyczne w ostrej białaczce, umożliwiające lepszą charakterystykę choroby czy też, jak w przypadku t(15;17), wybór celowanego leczenia9. Badanie to w każdym przypadku powinno być rozszerzone o badanie z użyciem techniki fluorescencyjnej hybrydyzacji in situ (FISH – fluorescence in situ hybridization), które umożliwia identyfikację chromosomów markerowych, aberracji liczbowych lub translokacji złożonych bądź ukrytych. Należy podkreślić, że nawet u 50-60% chorych w momencie rozpoznania stwierdza się różnego typu anomalie chromosomowe, mające charakter strukturalny bądź liczbowy10. Nawet u 10-20% pacjentów oceniany kariotyp zawiera co najmniej 3 zmiany chromosomalne (tzw. kariotyp złożony), natomiast u ok. 40% chorych na AML nie wykrywa się klasycznymi metodami żadnych zmian w kariotypie (tzw. kariotyp prawidłowy). Analiza materiału klinicznego dużych grup badawczych (np. SWOG/ECOG, MRC) umożliwiała podział pacjentów z AML na trzy grupy ryzyka cytogenetycznego w zależności od znaczenia prognostycznego stwierdzanych aberracji11,12. Szczegółowy opis zmian cytogenetycznych i molekularnych z podziałem na poszczególne grupy ryzyka przedstawiono w tabeli 2.
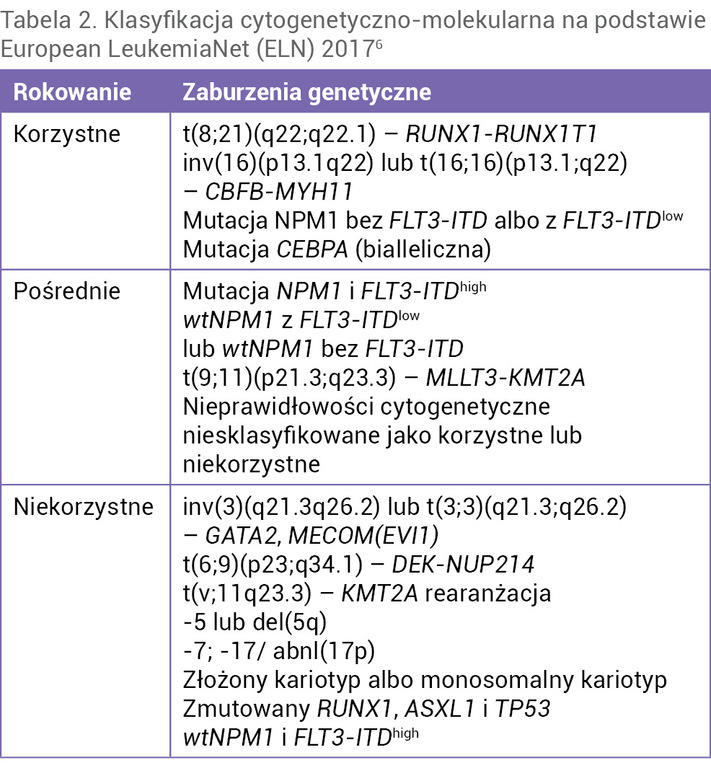
Tabela 2. Klasyfikacja cytogenetyczno-molekularna na podstawie European LeukemiaNet (ELN) 20176
Badania molekularne są obecnie złotym standardem w diagnostyce AML wykorzystywanym do szybkiego potwierdzenia obecności znanych genów fuzyjnych oraz identyfikacji istotnych rokowniczo mutacji somatycznych u pacjentów, u których w badaniu kariotypu nie stwierdza się dodatkowych aberracji. Gdy niezbędne jest podjęcie szybkiej decyzji dotyczącej wyboru terapii, badania te, szczególnie FLT3, BCR/ABL1 czy PML-RARA, powinny być wykonane w ciągu 2-3 dni, a pozostałe badania o znaczeniu rokowniczym, np. TP53, ASXL1, w ciągu pierwszego cyklu leczenia w celu jak najlepszej i najszybszej optymalizacji leczenia, w tym poszukiwania dawcy rodzinnego lub niespokrewnionego w przypadku identyfikacji czynników złego rokowania2,13,14.