


Spis treści
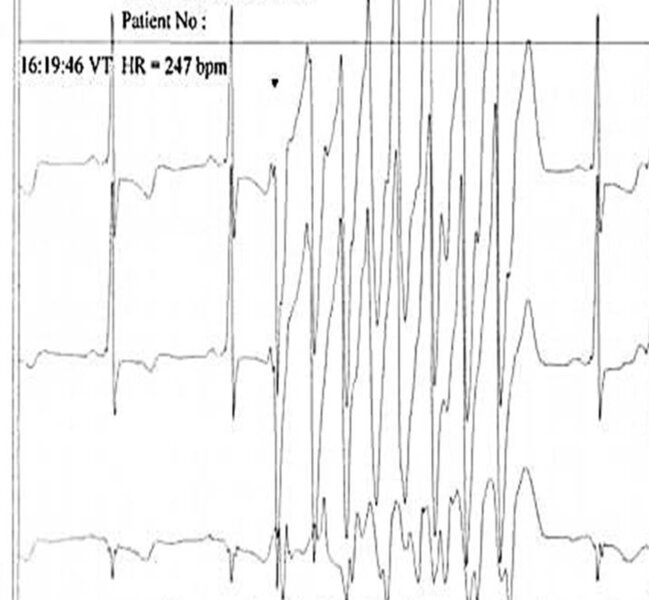
Rycina 1. Samoustępujący, szybki, wielokształtny częstoskurcz komorowy.
Wprowadzenie
Kanałopatie to genetycznie uwarunkowane choroby kanałów jonowych, występujące u osób bez choroby strukturalnej serca i odpowiedzialne za zagrażające życiu komorowe zaburzenia rytmu serca 1, 2 . Większość z nich odkryto niedawno, a wiedza na temat podłoża genetycznego kanałopatii jest jeszcze nowsza i nadal niepełna. Zespół długiego QT opisano po raz pierwszy w 1957 r. (pierwszy defekt genetyczny wykryto
 Dodaj do ulubionych
Dodaj do ulubionych
