U części chorych z RCM dużą rolę w ustaleniu potencjalnego czynnika etiologicznego odgrywa biopsja endomiokardialna17. Pozwala ona przede wszystkim na potwierdzenie lub wykluczenie zmian naciekowych oraz spichrzeniowych serca. Do pierwszej grupy należy gromadzenie się złogów amyloidu (amyloidoza serca) oraz żelaza (hemochromatoza). Drugi typ zmian obejmuje przede wszystkim choroby spichrzeniowe glikogenu i związków lipidowych18. W przypadku amyloidozy biopsja endomiokardialna pozwala nie tylko potwierdzić występowanie samej choroby, lecz także określić typ amyloidozy, co decyduje o włączeniu odpowiedniej terapii enzymatycznej (ryc. 10). Obraz histopatologiczny RCM charakteryzuje się swoistym włóknieniem śródmiąższowym. W przekroju poprzecznym można zauważyć obrączkowate obwódki dookoła kardiomiocytów3.
Arytmogenna kardiomiopatia prawokomorowa
Arytmogenna kardiomiopatia prawokomorowa jest chorobą dziedziczoną autosomalnie dominująco, w której dochodzi do zastępowania włókien mięśniowych serca tkanką włóknisto-tłuszczową, co w efekcie powoduje arytmie komorowe. Zmiany zachodzące w mięśniu sercowym rozwijają się stopniowo. Pierwsze zmiany następują w warstwie nasierdziowej, a ostatecznie dotyczą wszystkich warstw mięśnia sercowego19. Choroba najczęściej występuje u młodych osób, a pierwszym objawem może być krótkotrwała utrata przytomności związana z wysiłkiem fizycznym18. Chorzy oprócz zagrażających życiu arytmii komorowych mają również objawy niewydolności serca prawokomorowej19. Poza takimi objawami, jak kołatanie serca czy zawroty głowy, może również dojść do nagłego zatrzymania krążenia w przebiegu zaburzeń rytmu pracy komór, które najczęściej występują u młodych sportowców19. Na podstawie International Task Force 2010, na które składa się zapis EKG, badanie echokardiograficzne, MR serca oraz biopsja endomiokardialna, można ustalić ostateczne rozpoznanie. W każdym z wcześniej wspomnianych badań ARVC ma charakterystyczne zmiany.
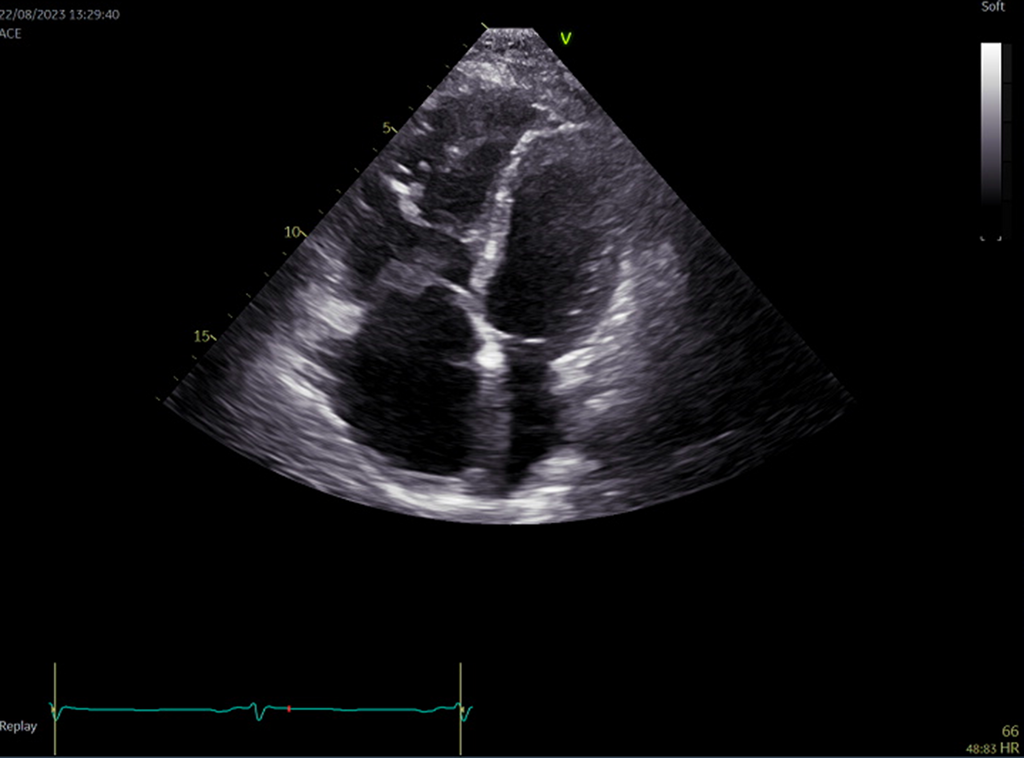
Rycina 11. Badanie echokardiograficzne. Arytmogenna kardiomiopatia prawej komory. Rozstrzeń prawej komory
W badaniu echokardiograficznym dla ARVC typowe są nieregularne poszerzenie drogi odpływu prawej komory oraz ścieńczenie grubości ściany prawej komory (ryc. 11). Można również zauważyć obszary dyskinetyczne, które wynikają z zastąpienia tkanki mięśniowej przez tkankę włóknisto-tłuszczową. Istotną cechą jest także obecność pojedynczych lub mnogich tętniaków19.
W MR serca obserwuje się odcinkową akinezę lub dyskinezę prawej komory, a ponadto dyssynchronię skurczu oraz jeden z następujących parametrów:
- stosunek objętości końcoworozkurczowej prawej komory (RV – right ventricle) do powierzchni ciała pacjenta (BSA – body surface area) od ≥100 do <110 ml/m2 (mężczyźni) lub od ≥90 do <100 ml/m2 (kobiety) albo
- frakcja wyrzutowa RV od >40% do ≤45%.
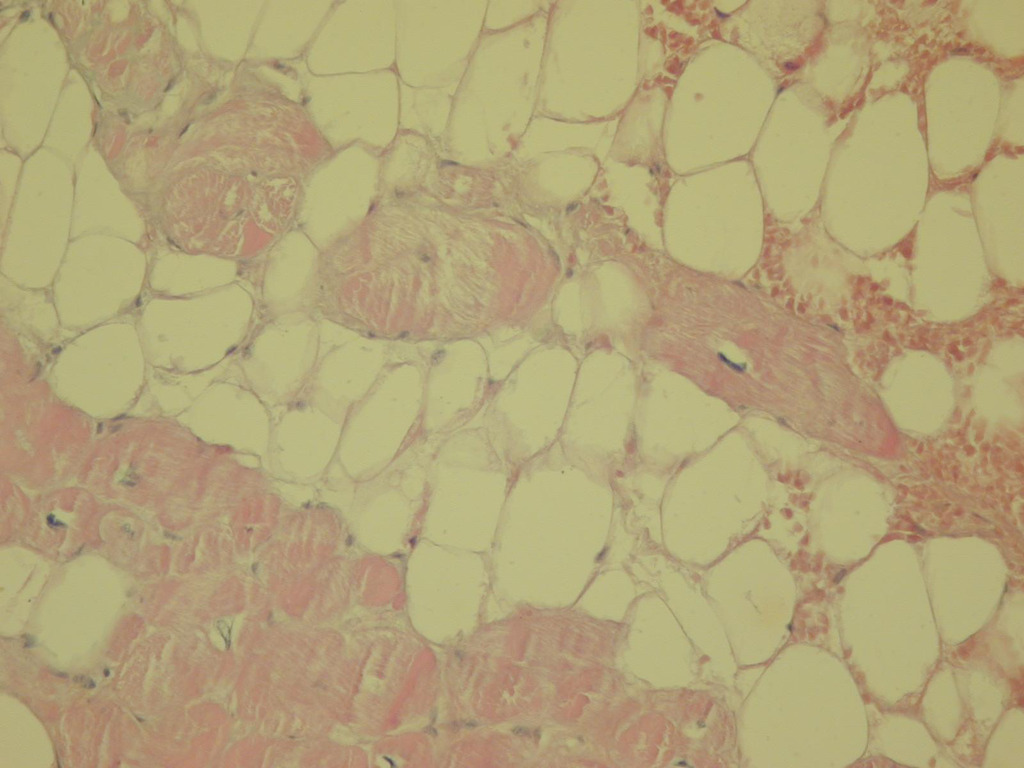
Rycina 12. Biopsja endomiokardialna. Arytmogenna kardiomiopatia prawej komory. Pomiędzy komórkami mięśniowymi obecna tkanka tłuszczowa. Barwienie histochemiczne (hematoksylina i eozyna)
W przypadku biopsji endomiokardialnej uważa się, że przynajmniej w jednym z pobranych wycinków z prawej komory tkanka mięśniowa serca musi być zastąpiona w >60% powierzchni skrawków tkanką włóknisto-tłuszczową (ryc. 12)3,18,19.
Nierozstrzeniowa kardiomiopatia lewej komory
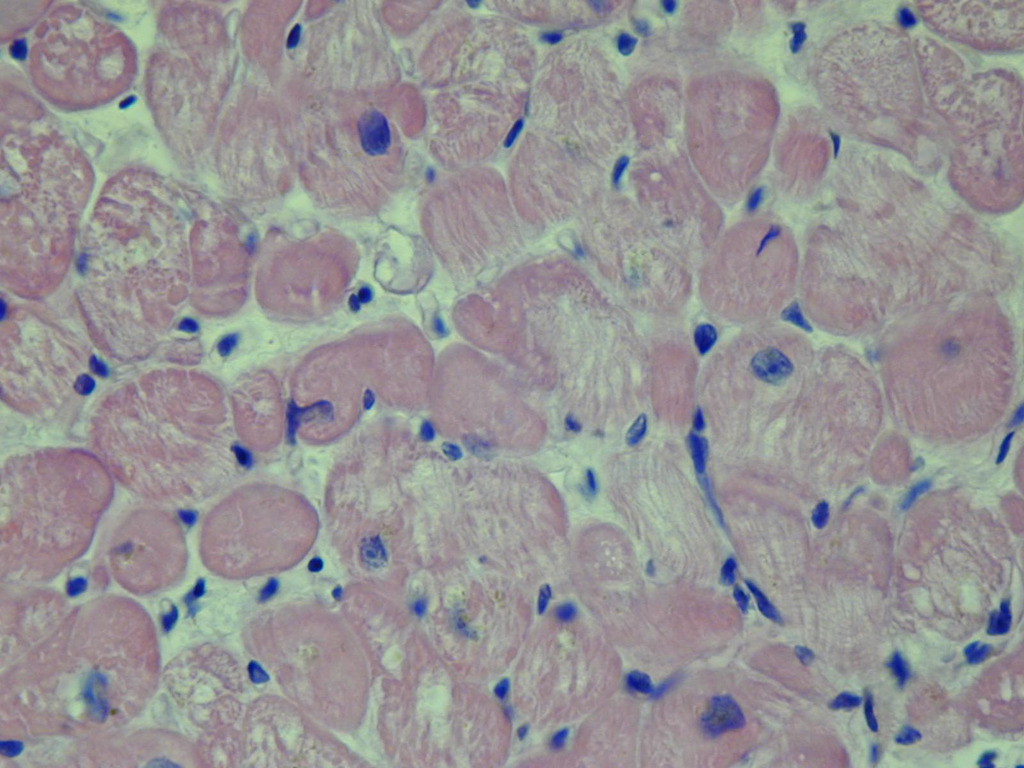
Rycina 13. Biopsja endomiokardialna. Nierozstrzeniowa kardiomiopatia lewej komory. Niespecyficzne zmiany wsteczne w kardiomiocytach w barwieniu hematoksyliną i eozyną
Nierozstrzeniowa kardiomiopatia lewej komory jest stosunkowo niedawno zaproponowaną grupą kardiomiopatii. W jej przebiegu chorzy mają zachowaną prawidłową funkcję skurczową. Jej cechą są zachowane wymiary jam serca z obecnością blizny o etiologii innej niż niedokrwienie czy też związaną z przebudową tłuszczową20. Sugeruje się, że nowo wyodrębniona grupa kliniczna powstała na potrzeby pacjentów, których przypadki zaklasyfikowano jako subkliniczna DCM lub potencjalnie rozwijająca się w kierunku DCM, niespełniających wszystkich diagnostycznych kryteriów21. Niektórzy nawet nazywają nierozstrzeniową kardiomiopatię lewej komory łagodniejszą wersją DCM, jednak ostatnie badania wykazały, że chorzy z NDLVC nie mogą być traktowani w kategorii łagodniejszej wersji choroby (ryc. 13)21. U chorych z NDLVC stwierdza się wyższe ryzyko wystąpienia migotania przedsionków, cukrzycy oraz hiperlipidemii20. Pomimo rosnącego zainteresowania tą jednostką chorobową nadal posiadamy bardzo mało informacji o jej przebiegu oraz rokowaniu pacjentów.
Podsumowanie
Pomimo rozwoju nowoczesnych technik diagnostycznych różnicowanie kardiomiopatii pozostaje wielkim wyzwaniem. Rozpoznanie kardiomiopatii jest przede wszystkim domeną badań klinicznych. Należy jednak podkreślić, że przyżyciowa diagnostyka morfologiczna serca wnosi dodatkowe, cenne informacje pozwalające ukierunkować postępowanie diagnostyczno-lecznicze. Dotyczy to w szczególności potwierdzenia lub wykluczenia podłoża zapalnego różnych schorzeń mięśnia sercowego. Ponadto biopsja mięśnia sercowego w wielu przypadkach pozwala na ostateczne ustalenie rozpoznania, tak jak w schorzeniach spichrzeniowych serca, co ma ogromne znaczenie w doborze odpowiedniej terapii.