Dzieci rodzą się zazwyczaj zdrowe, z donoszonej ciąży, mogą wystąpić trudności w adaptacji po urodzeniu, niższa może też być punktacja w skali Apgar. W momencie ujawnienia się drgawek większość pacjentów ma zaburzenia napięcia mięśniowego (hipotonia lub hipertonia), mogą występować drżenia i dystonie, rozdrażnienie, encefalopatia i nadmierna senność. Wśród ogólnych objawów najczęściej opisywane są zaburzenia karmienia, wymioty i zaburzenia oddychania7,10-12.
Nie ma stałego wzorca zapisu elektroencefalograficznego (EEG) charakterystycznego dla tego rodzaju padaczki – zazwyczaj są to zmiany wieloogniskowe, ale również może być zapis ze zmianami uogólnionymi czy nawet typu cisza–wyładowanie. W pojedynczych przypadkach na początku choroby zapis EEG był prawidłowy. W wyniku neuroobrazowania ośrodkowego układu nerwowego (OUN) może nie być żadnych zmian, czasami stwierdza się dysplazję lub hipoplazję ciała modzelowatego, wentrikulomegalię, nieprawidłowości w istocie białej, krwawienie do OUN, zmiany zanikowe, szczególnie w obszarze kory mózgu, czy ogniskową dysplazję korową7,10-12.
W podstawowych badaniach laboratoryjnych nie ma specjalnych odchyleń, czasami stwierdza się hipoglikemię i nieznacznie podwyższone stężenie kwasu mlekowego. Możliwe są zaburzenia w aminoacydogramie: w surowicy podwyższone stężenia treoniny, glicyny, tauryny i histydyny, a w płynie mózgowo-rdzeniowym (PMR) – poza ww. aminokwasami – również podwyższone stężenie glutaminianu, a obniżone PLP i GABA7,10-12.
Markerem biochemicznym choroby są metabolity, które powstają na drodze przemiany lizyny: Δ1P6C, AASA i kwas pipekolowy. Stężenia Δ1P6C i AASA pozostają podwyższone również po włączeniu leczenia witaminą B6, chociaż w mniejszym stopniu niż przed leczeniem. Stężenie kwasu pipekolowego może się unormować w trakcie leczenia, poza tym może być niespecyficznie podwyższone w niewydolności wątroby, a także w chorobach peroksysomalnych, z kolei stężenie AASA może być również podwyższone w deficycie kofaktora molibdenu. Niedawno zidentyfikowano nowy biomarker 6-oxo-pipekolan (6-oxo-PIP), który jest możliwy do wykrycia również z suchej kropli krwi na bibule (jest termostabilny) lub w porcji moczu, co stwarza szansę na populacyjne badania skriningowe w kierunku tej choroby w przyszłości13,14.
Obecnie zaleca się, aby przy podejrzeniu niedoboru antykwityny wykonać oznaczenie AASA i/lub Δ1P6C w surowicy, PMR lub moczu. Potwierdzenie choroby można również uzyskać na podstawie badania molekularnego9.
Postępowaniem z wyboru w przypadku stwierdzenia drgawek opornych na klasyczne leki przeciwpadaczkowe, szczególnie u noworodków i małych niemowląt, jest podaż witaminy B6. Pierwszą dawkę można podać dożylnie (100 mg), pamiętając, że u pacjentów z PDE-ALDH7A1 może dojść do bezdechu w trakcie podawania leku, dlatego należy taką próbę podejmować na oddziale intensywnej terapii z dostępem do respiratora. Następnie, niezależnie od odpowiedzi na podaż dożylną, należy kontynuować podaż witaminy B6 doustnie w dawce 30 mg/kg m.c./24 h w 3-4 dawkach podzielonych (maksymalnie 100-200 mg/24 h u noworodków i maksymalnie 300 mg/24 h u niemowląt). U dorosłych nie należy przekraczać dawki 500 mg/24 h (wyższe dawki mogą wywołać neuropatię). Są pacjenci, którzy odpowiadają na mniejsze dawki witaminy B6. Podawanie leku należy kontynuować do czasu wykluczenia biochemicznego i/lub molekularnego PDE-ALDH7A1, gdyż nie wszyscy chorzy odpowiadają na leczenie po pierwszych dawkach czy nawet dniach terapii. Zalecenia dotyczące leczenia PDE-ALDH7A1 wg rekomendacji z 2021 r. przedstawiono w tabeli 29.
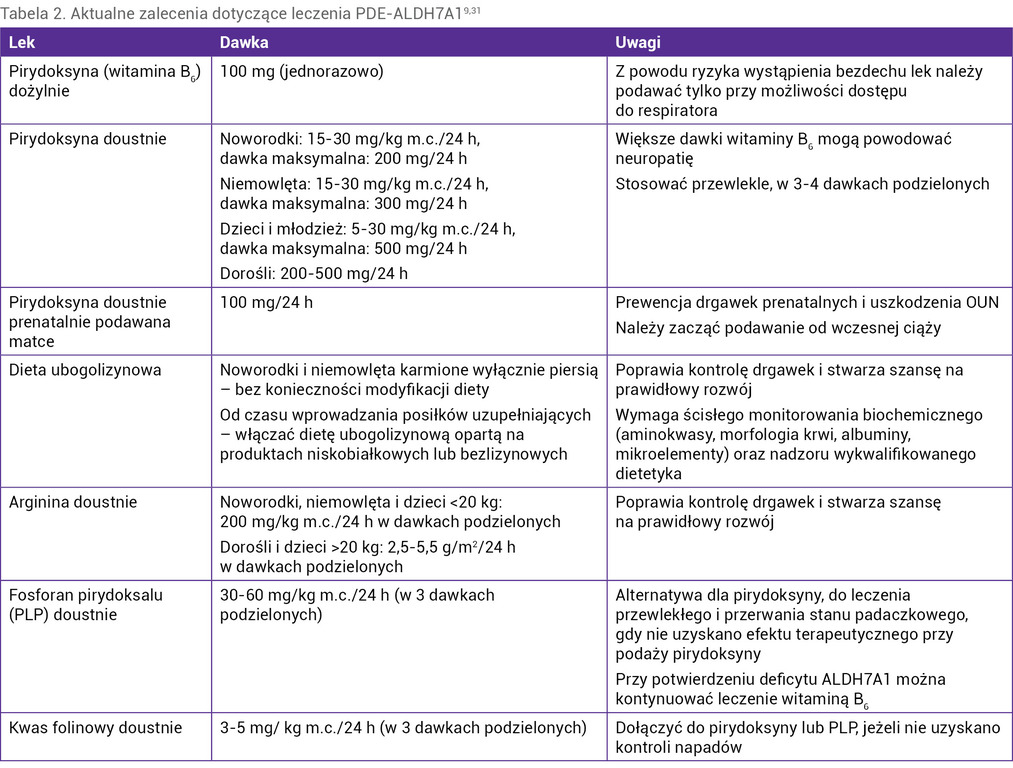
Tabela 2. Aktualne zalecenia dotyczące leczenia PDE-ALDH7A19,31
Jeżeli uda się uzyskać kontrolę drgawek po podaży witaminy B6, a nie potwierdzi się deficyt antykwityny, w diagnostyce różnicowej należy uwzględnić inne choroby zależne od witaminy B6 (tab. 1). W przypadku braku odpowiedzi na podaż witaminy B6 należy rozważyć inne wrodzone wady metabolizmu przebiegające z uporczywymi napadami, szczególnie te, które można leczyć lub ograniczyć ich postęp i objawy kliniczne specyficznym postępowaniem (tab. 3 i 4)15-17.

Tabela 3. Możliwe leczenie w wybranych wrodzonych wadach metabolizmu przebiegających z drgawkami15-17

Tabela 4. Wrodzone wady metabolizmu przebiegające z drgawkami ujawniające się w różnych okresach życia15-17
U 90% pacjentów z PDE-ALDH7A1 udaje się uzyskać kontrolę napadów po podaży samej witaminy B6, jednak mimo to u 75% chorych stwierdza się opóźnienie rozwoju lub niepełnosprawność intelektualną w różnym stopniu. Okazuje się, że ograniczenie podaży lizyny, a tym samym obniżenie stężenia w płynach ustrojowych m.in. AASA czy Δ1P6C, stwarza możliwość poprawy rozwoju poznawczego w tej grupie pacjentów. Podobnie działa suplementacja argininy, aminokwasu, który z lizyną konkuruje o transporter w jelitach i przy przechodzeniu przez barierę krew–mózg w OUN18-23. Pokarm kobiecy zawiera niewiele lizyny, dlatego noworodki i małe niemowlęta mogą być karmione mlekiem matki, a w okresie rozszerzania diety zaleca się produkty niskobiałkowe i preparaty bez lizyny (produkty spożywcze specjalnego przeznaczenia). U starszych dzieci i dorosłych w przypadku braku dostępności produktów ubogo- lub bezlizynowych można stosować dietę niskobiałkową. Należy jednak pamiętać, że dieta nie może być deficytowa i trzeba ją prowadzić pod kontrolą wykwalifikowanego dietetyka, z regularną kontrolą kliniczną i biochemiczną. Stężenie lizyny w surowicy powinno być utrzymywane w dolnych granicach wartości referencyjnej dla wieku9.
Suplementacja argininy u pacjentów z PDE-ALDH7A1 powinna wynosić 200 mg/kg m.c./24 h w dawkach podzielonych, dawka maksymalna u dorosłego to 4-5,5 g/m2/24 h9.
Opisano pojedyncze przypadki chorych z potwierdzonym biochemicznie i molekularnie deficytem antykwityny, którzy nie odpowiedzieli na terapeutyczne dawki witaminy B6, natomiast drgawki ustąpiły u nich po podaży kwasu folinowego. U chorych tych w chromatografii PMR stwierdzono niezidentyfikowany dotychczas pik substancji X. Dlatego przy uporczywych drgawkach u noworodków i małych dzieci, które nie odpowiedziały na leczenie witaminą B6, należy dołączyć kwas folinowy w dawce 3-5 mg/kg m.c./24 h w 2 dawkach podzielonych3.
Deficyt oksydazy fosforanu pirydoksaminy
Oksydaza fosforanu pirydoksaminy (PNPO – pyridoxamine phosphate oxidase) jest enzymem niezbędnym do powstania aktywnej formy witaminy B6, czyli PLP. Enzym ten kodowany jest przez gen PNPO (opisano 28 patogennych wariantów), a choroba dziedziczy się autosomalnie recesywnie. Jego deficyt (OMIN#6032870) prowadzi do encefalopatii padaczkowej, ujawniającej się od pierwszych dni życia24.
Obraz kliniczny jest bardzo podobny do obrazu występującego w przypadku deficytu antykwityny, uważa się jednak, że przebieg kliniczny jest cięższy niż w PDE-ALDH7A1. Dodatkowo ok. 40% pacjentów odpowiada redukcją lub ustąpieniem napadów po podaży samej witaminy B6. W badaniach biochemicznych może wystąpić hipoglikemia, kwasica metaboliczna i podwyższone stężenie kwasu mlekowego oraz treoniny i glicyny. W moczu stwierdza się wydalanie kwasu wanilinomlekowego (VLA – vanillin lactic acid) i kwasu homowanilinowego (HVA – homovanillic acid), w PMR zaś obniżone jest stężenia kwasu hydroksyindolooctowego (HIAA – hydroxyindoleacetic acid), HVA, pirydoksyny i PLP oraz argininy. Nie ma specyficznego biomarkera choroby, a obniżenie stężenie PLP w PMR zawsze powinno sugerować drgawki pirydoksynozależne. Potwierdzeniem choroby jest oznaczenie aktywności enzymu PNPO w suchej kropli krwi3.
Szybkie włączenie leczenia fosforanem pirydoksalu (3-60 mg/kg m.c./24 h) stwarza szansę na prawidłowy rozwój dziecka25. Dotychczas opisano w piśmiennictwie nieco ponad 60 pacjentów z potwierdzonym deficytem PNPO3.