



Co znajdziesz w artykule?
Ostatnie lata przyniosły znaczny postęp w badaniach nad molekularnymi i immunologicznymi aspektami karcynogenezy. Nadal trwają poszukiwania kolejnych parametrów, które z większą precyzją charakteryzowałyby nowotwory i pomagałyby określać rokowanie u chorych. Jednym z nowych parametrów, który znajduje się w kręgu zainteresowań badaczy, jest związany z czynnikiem martwicy nowotworu ligand indukujący apoptozę (tumor necrosis factor-related apoptosis-inducing ligand – TRAIL).
Spis treści
APOPTOZA
Apoptoza, nazywana również programowaną śmiercią komórki, jest procesem fizjologicznym uwarunkowanym genetycznie, polegającym na zmianach biochemicznych i morfologicznych komórek w efekcie proteolitycznej i nukleolitycznej degradacji ich składników, co w konsekwencji prowadzi do śmierci tych komórek. 1, 2, 3 W warunkach fizjologicznych prawidłowo przebiegająca apoptoza jest niezbędna do utrzymania prawidłowej homeostazy tkankowej. 1 Jej zaburzenia odgrywają istotną rolę w
powstawaniu oraz progresji nowotworów, prowadząc do nieprawidłowego zwiększania się żywotności komórek, wydłużania ich życia i utrwalania już zaistniałych mutacji. 2, 4 Apoptoza może być inicjowana dwiema drogami: wewnątrzkomórkową i zewnątrzkomórkową, które nierzadko ze sobą współwystępują. 2, 5
Droga wewnątrzpochodna apoptozy, związana z mitochondriami i z aktywacją genów rodziny bcl-2, jest typową reakcją komórki na uszkodzenie DNA lub inne bodźce wewnątrzkomórkowe. 2, 4 W ten sposób przebiega zależna od genu p53 apoptoza komórkowa będąca skutkiem chemioterapii i radioterapii. 1, 2, 4, 6 Wiele nowotworów unika jednak apoptozy wewnątrzpochodnej na drodze mutacji i dezaktywacji genu p53. 4
W mechanizmie zewnątrzpochodnym apoptoza jest indukowana w sposób niezależny od genu p53, poprzez przyłączenie się ligandu do tzw. receptorów śmierci. 4, 6 W zainicjowaniu tego procesu mają udział komórki immunokompetentne. Mogą one działać przez uwolnienie perforyn i granzymów lub na drodze interakcji z ligandami rodziny czynnika martwicy nowotworu (tumor necrosis factor – TNF): TNF-α, FasL (CD95L) i TRAIL, które są związane z „receptorami śmierci” na komórkach docelowych. 1, 2, 3, 4 W efekcie pobudzenia „receptorów śmierci” dochodzi do kaskady zdarzeń komórkowych prowadzących do zmniejszania się komórki, rozpadu jądra i powstania ciałek apoptotycznych, usuwanych następnie przez inne komórki na drodze fagocytozy. 2, 4, 6
LIGAND TRAIL
Do rodziny TNF należy TRAIL – białko błonowe typu II, zbudowane z 281 aminokwasów, opisane po raz pierwszy w 1995 roku przez Wiley i wsp. 7 TRAIL jest kodowany przez gen zlokalizowany na chromosomie 3, składający się z pięciu egzonów i czterech intronów. 7 Wykazuje on sekwencje homologiczne z innymi białkami rodziny TNF, jak Fas (CD95) i RNF, odpowiednio w 28 proc. i 33 proc. 7 Ligand TRAIL występujący w postaci rozpuszczalnej ma masę 20 kDa, natomiast w formie związanej z powierzchnią komórek immunokompetentnych, takich jak komórki dendrytyczne, makrofagi, limfocyty T i komórki NK, masę 33 kDa. 8, 9 Ligand TRAIL i jego receptory są obecne w wielu prawidłowych tkankach organizmu człowieka. 7, 8, 10
W przeciwieństwie do innych ligandów, jak TNF-α czy FasL, których pobudzenie jest toksyczne dla prawidłowych komórek organizmu, TRAIL ma zdolność indukowania apoptozy w komórkach nowotworowych, nie wykazując jednocześnie cytotoksyczności w stosunku do komórek prawidłowych, co potwierdzono w badaniach in vitro oraz in vivo. 3, 7, 8, 11, 12, 13 W wywołaniu apoptozy indukowanej przez TRAIL w komórkach nowotworów mogą pomagać czynniki chemiczne i fizyczne działające w mechanizmie wewnątrzpochodnym. 3, 14, 15, 16 Czynnikiem przeciwdziałającym apoptozie indukowanej przez TRAIL jest natomiast niedotlenienie, obecne często w centralnych fragmentach szybko rosnących nowotworów. 3 Obserwacja, że TRAIL selektywnie indukuje apoptozę jedynie w komórkach nowotworowych, nie powodując podobnego zjawiska w komórkach niezmienionych nowotworowo, stała się podstawą do licznych opracowań naukowych. 12, 13, 17, 18, 19, 20, 21
RECEPTORY DLA LIGANDU TRAIL
Ligand TRAIL działa poprzez cztery receptory TRAIL-R1 (DR4), TRAIL-R2 (DR5), TRAIL-R3 (DcR1/TRID/LIT) oraz TRAIL-R4 (DcR2/TRUNDD) znajdujące się na powierzchni komórek. Piątym receptorem jest rozpuszczalna osteoprotegryna (OPG), a połączenie z nią TRAIL nie wywołuje apoptozy. 9, 10, 11, 21, 22, 23, 24, 25 Wszystkie receptory komórkowe dla TRAIL mają sekwencje homologiczne w swojej domenie zewnątrzkomórkowej. Wewnątrzkomórkowe domeny receptorów DR4 i DR5 po połączeniu receptora z ligandem pełnią podstawową rolę dla zapoczątkowania apoptozy. 8, 21, 22, 23, 24, 25 Potwierdzono wysokie podobieństwo sekwencji receptorów DR4 i DR5 i różnice w termodynamice połączeń obu tych receptorów z ligandem TRAIL. 25
Obecnie uważa się, że pobudzenie „receptorów śmierci” DR4 i DR5 powoduje ich oligomeryzację i indukuje apoptozę poprzez wewnątrzkomórkową domenę śmierci (death domain – DD). Wskutek połączenia DD z białkiem adaptorowym FADD (Fas-associated death domain) oraz z cytoplazmatyczną prokaspazą 8 powstaje kompleks DISC (death inducing signaling complex). 1, 2, 4, 8, 24 Połączenie DD z białkiem adaptorowym FADD może również aktywować prokaspazę 10, która prawdopodobnie wpływa na efekt proliferacyjny „receptorów śmierci”. 19 W niektórych komórkach już sama aktywacja kaspazy 8 jest wystarczająca do dalszego przebiegu apoptozy, w części komórek niezbędna jest również aktywacja drogi wewnątrzpochodnej. Aktywacja prokaspazy 8 prowadzi do aktywacji kaspazy 3, wiodąc w konsekwencji do wyzwolenia kaskady procesów wewnątrzkomórkowych prowadzących do oddzielenia się komórki od innych komórek, utraty wody i jej obkurczenia się. 1, 8, 9, 21, 26 Na różnych etapach dochodzi do aktywacji kaspazy 6 i 9 oraz innych substancji (ryc. 1). 2, 8, 9, 26
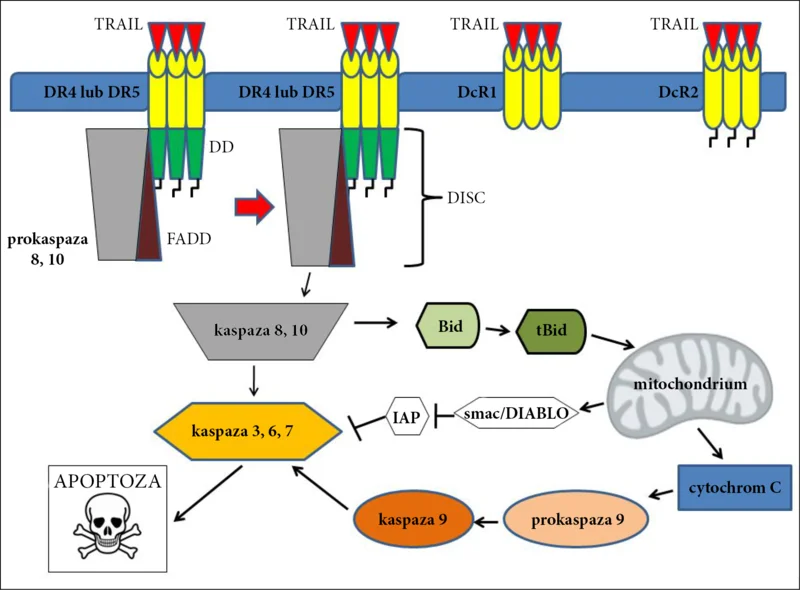
Ryc. 1. Schematyczne przedstawienie apoptozy indukowanej przez TRAIL.
Ekspresję obu receptorów proapoptotycznych, tj. DR4 i DR5, wykazano w wielu lokalizacjach, w tym w leukocytach obwodowych, hepatocytach, neuronach, kanalikach krętych nerki, komórkach mięśnia sercowego, komórkach nabłonka jelitowego, nabłonku oskrzeli, komórkach przegród pęcherzyków płucnych, komórkach Leydiga, endometrium, a także w komórkach zarodkowych. 10, 27, 28, 29 Ekspresję DR4 bez obecności ekspresji DR5 potwierdzono w nabłonku dróg żółciowych, a obecność DR5 bez obecności DR4 zaobserwowano w nabłonku naczyń mózgowych, w komórkach pętli Henlego, w blaszce właściwej błony śluzowej jelit i w nabłonku naczyń oskrzelowych. 10
Przyłączenie się ligandu TRAIL do receptorów DcR1 i DcR2 nieposiadających domen cytoplazmatycznych, w przeciwieństwie do pobudzenia receptorów DR4 i DR5, nie prowadzi do apoptozy. 8, 11 Dochodzi jedynie do związania cząsteczek ligandu, pośrednio więc do zmniejszenia wysycenia ligandem receptorów DR4 i DR5 na drodze konkurencji i w efekcie do przeciwdziałania apoptozie. 23 Jedna z koncepcji zakłada, że przyczyną, dla której ligand TRAIL nie powoduje apoptozy w prawidłowych komórkach organizmu, jest proporcjonalnie wysoka ekspresja na ich powierzchni „fałszywych” receptorów, tj. DcR1 i DcR2 („receptory ochronne”). 12 Wyniki badań ostatnich lat pokazują jednak, że nie jest to jedyny mechanizm wyjaśniający zjawisko oporności prawidłowych komórek na apoptozę indukowaną przez TRAIL. 17, 21, 30, 31
Obecność „receptorów ochronnych” DcR1 i DcR2 została potwierdzona w:
- hepatocytach,
- komórkach dróg żółciowych,
- nabłonku jelita grubego,
- neuronach,
- endometrium,
- komórkach mięśnia sercowego,
- komórkach zarodkowych,
- komórkach Leydiga. 10, 27, 32, 33
Ekspresję DcR2 przy braku ekspresji DcR1 znaleziono w:
- kanalikach krętych nerki,
- komórkach nabłonka jelitowego,
- komórkach krypt jelitowych,
- nabłonku oskrzeli,
- przegrodach między pęcherzykami płucnymi,
- nabłonku naczyń krwionośnych płuc. 10, 27
Ekspresję DcR1 bez obecności DcR2 stwierdzono natomiast w:
- nabłonku naczyniowym w mózgu,
- sercu. 10
LIGAND TRAIL JAKO POTENCJALNY ELEMENT W LECZENIU PRZECIWNOWOTWOROWYM
W ostatnich latach coraz częściej wskazuje się na rolę TRAIL jako cząsteczki efektorowej w zabijaniu komórek nowotworu złośliwego i jako potencjalnego narzędzia w leczeniu przeciwnowotworowym. 8, 34 Badania fazy przedklinicznej, a także badania kliniczne z zastosowaniem rekombinowanego ludzkiego ligandu TRAIL (recombinant human TRAWL – rhTRAIL) oraz agonistów receptora dla TRAIL w licznych nowotworach wykazały korzystny efekt przeciwnowotworowy takiego postępowania. 5, 9 Potencjał terapeutyczny rozpuszczalnego rekombinowanego TRAIL potwierdzono m.in. w:
- płaskonabłonkowym raku głowy i szyi,
- raku piersi,
- raku płuc,
- raku jelita grubego,
- raku odbytnicy,
- raku trzustki,
- raku nerki,
- raku tarczycy,
- raku endometrium,
- raku gruczołu krokowego,
- glejakach,
- szpiczaku mnogim,
- białaczkach. 16, 20, 21, 34, 35
W piśmiennictwie spotyka się jednak także doniesienia, które nie potwierdzają tej zależności, wskazując na oporność niektórych nowotworów na TRAIL, w tym części linii raka piersi, raka płuc (NSCLC), raka nerki, nowotworów układu krwiotwórczego (przewlekła białaczka limfocytarna) i guzów ośrodkowego układu nerwowego (niektóre glejaki, oponiaki, rdzeniak zarodkowy). 5, 30, 35, 36, 37
Badania ostatnich lat pokazują, że stosując odpowiednie chemioterapeutyki, można „uwrażliwić” na TRAIL komórki nowotworowe pierwotnie na niego niewrażliwe. 9, 16, 18, 21, 36, 38, 39 Zjawisko takie zaobserwowano, m.in. stosując sorafemib w badaniach nad rakiem endometrium. 15 Podobne wyniki uzyskano w badaniach nad rakiem jajnika i szyjki macicy oraz w nowotworach złośliwych w innych lokalizacjach. 12, 15, 40, 41
Są także prowadzone badania w celu wykorzystania rekombinowanego ligandu TRAIL jako radiouczulacza u chorych poddawanych radioterapii w celu zwiększenia promienioczułości nowotworu. 14 W badaniach in vitro stwierdzono bowiem zwiększenie promienioczułości w pierwotnie niewrażliwych na radioterapię komórkach HeLa raka szyjki macicy oraz raka piersi, płuca, jelita grubego, a także nowotworu głowy i szyi, gdy pobudzono ich proapoptotyczne receptory dla TRAIL. 42, 43
Możliwości i warunki do zastosowania ligandu TRAIL w praktyce klinicznej powinny być jednak rozpatrywane ostrożnie, ze względu na prawdopodobne niekorzystne działanie na ludzkie hepatocyty, szczególnie u pacjentów z zaburzeniami funkcji wątroby. 16, 44, 45
ROZMIESZCZENIE I MOŻLIWE ZNACZENIE KLINICZNE RECEPTORÓW DLA TRAIL W NOWOTWORACH ZŁOŚLIWYCH
Większość opracowań dotyczących receptorów dla ligandu TRAIL w nowotworach dotyczy ich ekspresji cytoplazmatycznej. Opisano ją w wielu liniach komórkowych nowotworów in vitro, ale także w tkankach nowotworowych in vivo. Ekspresję cytoplazmatyczną DR4 i DR5 stwierdzono w:
- pierwotnych i przerzutowych guzach mózgu,
- białaczkach,
- czerniaku skóry,
- raku płuc,
- raku piersi,
- raku głowy i szyi,
- raku przełyku,
- raku jelita grubego,
- raku odbytnicy,
- raku trzustki,
- raku nerki,
- raku pęcherza moczowego,
- raku szyjki macicy,
- raku jajnika. 27, 46, 47, 48, 49, 50
Wskazuje się na fakt, że występowanie błonowej ekspresji DR4 i DR 5 nie jest częste w przypadku ich wysokiej ekspresji cytoplazmatycznej. Taką właśnie zależność opisali Ganten i wsp. w raku piersi oraz Younes i wsp. w raku przełyku. 46, 48 W badaniu Gottwalda i wsp. błonową ekspresję DR4 i DR5 w gruczolakoraku endometrioidalnym endometrium obserwowano rzadko, odpowiednio u 12 proc. i 4 proc. chorych. 29 Podobnie niskie odsetki przypadków wykazujących ekspresję błonową receptorów DR4 i DR5 opisano już wcześniej w rakach piersi i przełyku. 46, 48
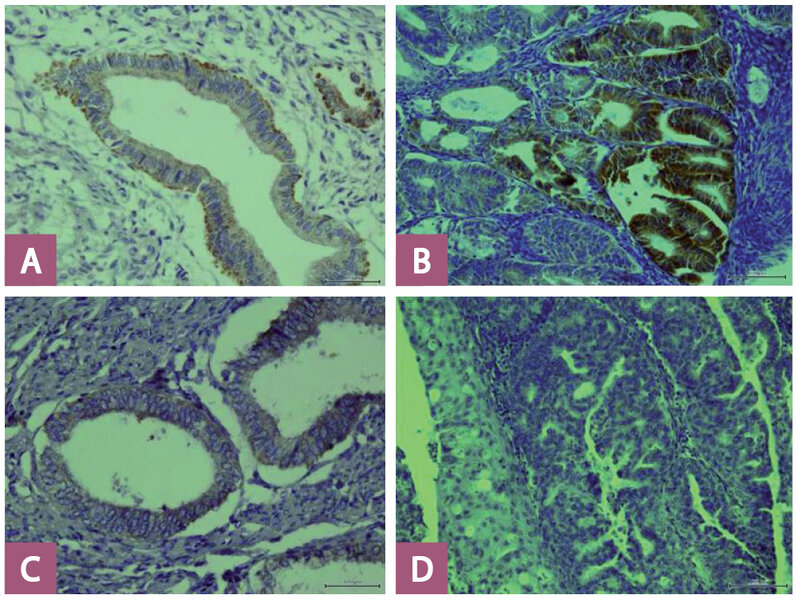
Ryc. 2. A – obecna ekspresja błonowa DR4 w prawidłowym endometrium (powiększenie 400 ×); B – obecna ekspresja błonowa DR4 w raku endometrium (powiększenie 400 ×); C – obecna ekspresja błonowa DR5 w prawidłowym endometrium (powiększenie 400 ×); D – brak ekspresji błonowej DR5 w raku endometrium (powiększenie 400 ×).
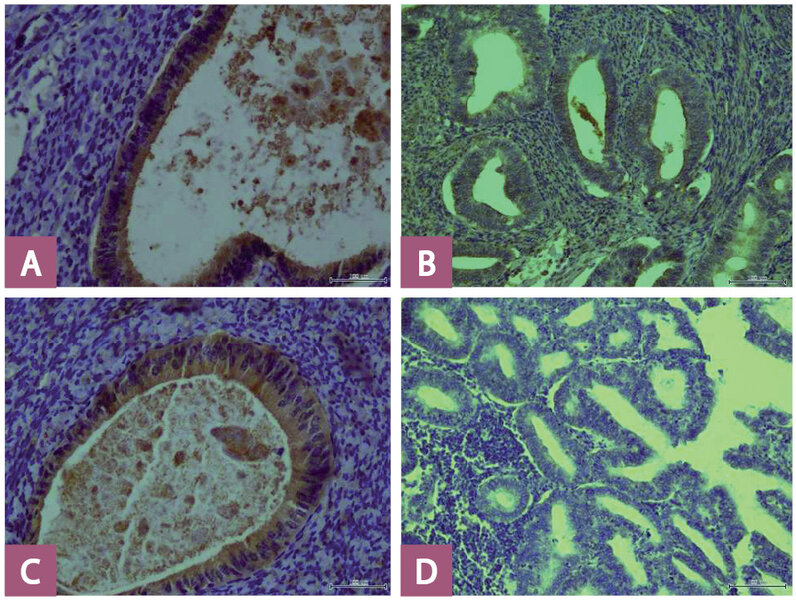
Ryc. 3. A – obecna ekspresja błonowa DcR1 w prawidłowym endometrium (powiększenie 400 ×); B – obecna ekspresja błonowa DcR1 w raku endometrium (powiększenie 400 ×); C – obecna ekspresja błonowa DcR2 w prawidłowym endometrium (powiększenie 400 ×); D – brak ekspresji błonowej DcR2 w raku endometrium (powiększenie 400 ×).
W badaniach prowadzonych na tkankach nowotworowych również oceniano ekspresję „receptorów ochronnych” dla ligandu TRAIL. Frank i wsp. ocenili ekspresję cytoplazmatyczną DcR1 i DcR2 w 19 pierwotnych i 11 przerzutowych guzach mózgu, stwierdzając obecność DcR1 odpowiednio w 18/19 i 9/11 przypadków, a DcR2 w 6/19 i 1/11 przypadków. 27 W tkankach raka endometrium cytoplazmatyczna ekspresja DcR1, bez jednoczesnej oceny pozostałych receptorów dla TRAIL, została opisana przez Tarragonę i wsp. w 98 proc. z analizowanych 62 przypadków raka endometrium. 32 W badaniu Gottwalda i wsp. błonowa ekspresja DcR1 i DcR2 była obecna odpowiednio u 27,3 proc. i u 18,7 proc. chorych na raka endometrium. 33 Połączenie wyników obu tych badań daje obraz rzeczywistego rozkładu „receptorów ochronnych” dla ligandu TRAIL w komórkach raka endometrium.
Z klinicznego punktu widzenia znajomość występowania i rozmieszczenia receptorów dla TRAIL w komórkach i tkankach nowotworów może okazać się istotna. Pojawiły się bowiem doniesienia o występowaniu korelacji wielkości ekspresji cytoplazmatycznej obydwu „receptorów śmierci” oraz „receptorów ochronnych” dla ligandu TRAIL ze zróżnicowaniem histologicznym i zawansowaniem nowotworów. 46 Koksal i wsp. w raku gruczołu krokowego stwierdzili również dodatnią korelację ekspresji DcR2 z punktacją w skali Gleasona oraz ze stężeniem PSA. 54 Nie wszyscy autorzy potwierdzają jednak występowanie podobnych zależności. Koornstra i wsp. nie zaobserwowali zależności pomiędzy ekspresją cytoplazmatyczną DR4 i DR5 a zróżnicowaniem histologicznym i zaawansowaniem raka okrężnicy. 27 Z kolei u chorych z rakiem endometrium Tarragona i wsp. opisali brak różnic w obecności i wielkości ekspresji DcR1 w zależności od typu i zróżnicowania histologicznego oraz zaawansowania nowotworu. 32
W piśmiennictwie pojawiają się również prace mówiące o możliwym znaczeniu prognostycznym ekspresji receptorów dla TRAIL w chorobie nowotworowej. Poglądy badaczy w tym zakresie różnią się jednak znacząco. Li i wsp. w dziesięcioletniej obserwacji opisali istotnie dłuższe przeżycie bez wznowy u chorych z rakiem pęcherza moczowego z wysoką ekspresją DR4 i DR5 w porównaniu z chorymi z niską ekspresją obydwu tych receptorów. 47 W raku gruczołu krokowego Koksal i wsp. opisali zależność pomiędzy wysoką ekspresją DcR2 a wystąpieniem wznowy i długością przeżycia. 54 Z drugiej jednak strony Zhuang i wsp. nie stwierdzili zależności cytoplazmatycznej ekspresji DcR1 i DcR2 z rokowaniem w chorobie nowotworowej. 55
PODSUMOWANIE
1. Chociaż funkcje i znaczenie ligandu TRAIL, jego „rceptorów śmierci” i „receptorów ochronnych” oraz osteoprotegryny w prawidłowych i zmienionych nowotworowo komórkach są jest już dość dobrze zdefiniowane, to jednak wymagają dalszych badań.
2. Heterogenność receptorów sugeruje złożoność mechanizmów związanych z ich biologiczną funkcją, a wielkość ich ekspresji nie musi korelować z odpowiedzią na ligand TRAIL.
3. Należy mieć nadzieję, że dokonujący się na naszych oczach olbrzymi postęp pozwoli w niedługim czasie jednoznacznie określić możliwości zastosowania ligandu TRAIL i jego receptorów w praktyce klinicznej.
- 1. Portt L, Norman G, Clapp C et al. Anti-apoptosis and cell survival: a review. Biochim Biophys Acta 2011;1813:238-59
- 2. Wyllie AH. Where, O death, is thy sting? A brief review of apoptosis biology. Mol Neurobiol 2010;42:4-9
- 3. Park SY, Billiar TR, Seol DW. Hypoxia inhibition of apoptosis induced by tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). Biochem Biophys Res Commun 2002;291:150-3
- 4. Hotchkiss RS, Strasser A, McDunn JE et al. Cell death. N Engl J Med 2009;361:1570-83
- 5. Zhao J, Lu Y, Shen HM. Targeting p53 as a therapeutic strategy in sensitizing TRAIL-induced apoptosis in cancer cells. Cancer Lett 2012;314:8-23
- 6. Helewski KJ, Kowalczyk-Ziomek GI, Konecki J. Apoptoza i martwica – dwie drogi do jednego celu. Wiad Lek 2006;59:679-84
- 7. Wiley SR, Schooley K, Smolak PJ et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 1995;3:673-82
- 8. Mellier G, Huang S, Shenoy K et al. TRAILing death in cancer. Mol Aspects Med 2010;31:93-112
- 9. Pennarun B, Meijer A, de Vries EG et al. Playing the DISC: turning on TRAIL death receptor-mediated apoptosis in cancer. Biochim Biophys Acta 2010;1805:123-40
- 10. Spierings DC, de Vries EG, Vellenga E et al. Tissue distribution of the death ligand TRAIL and its receptors. J Histochem Cytochem 2004;52:821-31
- 11. Gura T. How TRAIL kills cancer cells, but not normal cells. Science 1997;277:768
- 12. Smyth MJ, Takeda K, Hayakawa Y et al. Nature’s TRAIL-on path to cancer immunotherapy. Immunity 2003;18:1-6
- 13. Kendrick JE, Estes JM, Straughn Jr. JM et al. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and its therapeutic potential in breast and gynecologic cancers. Gynecol Oncol 2007;106:614-21
- 14. Niemoeller OM, Belka C. Radiotherapy and TRAIL for cancer therapy. Cancer Lett 2013;332:184-93
- 15. Llobet D, Eritja N, Yeramian A et al. The multikinaze inhibitor Sorafenib induces apoptosis and sensitizes endometrial cancer cells to TRAIL by different mechanisms. Eur J Cancer 2010;46:836-50
- 16. Wu X, Hui KM. Induction of potent TRAIL-mediated tumoricidal activity by hFLEX/Furin/TRAIL recombinant DNA construct. Mol Ther 2004;9:674-81
- 17. Wu G. TRAIL as a target in anti-cancer therapy. Cancer Lett 2009;285:1-5
- 18. Maksimovic-Ivanic D, Stosic-Grujicic S, Nicoletti F et al. Resistance to TRAIL and how to surmount it. Immunol Res 2012;52:157-68
- 19. Kruyt F. TRAIL and cancer therapy. Cancer Lett 2008;263:14-25
- 20. Raulf N, El-Attar R, Kulms D et al. Differential response of head and neck cancer cell lines to TRAIL or Smac mimetics is associated with the cellular levels and activity of caspase-8 and caspase-10. Br J Cancer 2014;111:1955-64
- 21. Amm HM, Oliver PG, Lee CH et al. Combined modality therapy with TRAIL or agonistic death receptor antibodies. Cancer Biol Ther 2011;11:431-49
- 22. Pan G, Ni J, Wei YF et al. An antagonist decoy receptor and a death domain-containing receptor for TRAIL. Science 1997;277:215-8
- 23. Sheridan JP, Marsters SA, Pitti RM et al. Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science 1997;277:818-27
- 24. van Roosmalen IA, Quax WJ, Kruyt FA. Two death-inducing human TRAIL receptors to target in cancer: similar or distinct regulation and function? Biochem Pharmacol 2014;91:447-56
- 25. Ramamurthy V, Yamniuk AP, Lawrence EJ et al. The structure of the death receptor 4-TNF-related apoptosis-inducing ligand (DR4-TRAIL) complex. Acta Crystallogr F Struct Biol Commun 2015;71:1273-81
- 26. Slee EA, Adrain C, Martin SJ. Executioner caspase-3, -6, and -7 perform distinct, non-redundant roles during the demolition phase of apoptosis. J Biol Chem 2001;276:7320-6
- 27. Koornstra JJ, Kleibeuker JH, van Geelen C et al. Expression of TRAIL (TNF-related apoptosis-inducing ligand) and its receptors in normal colonic mucosa, adenomas and carcinomas. J Pathol 2003;200:327-35
- 28. Frank S, Köhler U, Schackert G et al. Expression of TRAIL and its receptors in human brain tumors. Biochem Biophys Res Comm 1999;257:454-9
- 29. Gottwald L, Szwalski J, Piekarski J et al. Membrane expression of the death ligand TRAIL receptors DR4 and DR5 in the normal endometrium, endometrial atypical hyperplasia and endometrioid endometrial cancer. J Obstet Gynaecol 2013;33:512-8
- 30. Stegehuis JH, de Wilt LH, de Vries EG et al. TRAIL receptor targeting therapies for non-small cell lung cancer: current status and perspectives. Drug Resist Updat 2010;13:2-15
- 31. Wang D, Lu J, Tindall DJ. Androgens regulate TRAIL-induced cell death in prostate cancer cells via multiple mechanisms. Cancer Lett 2013;335:136-44
- 32. Tarragona J, Llecha N, Santacana M, Lopez S, Gatius S, Llobet D, Dolcet X, Palomar-Asenjo V, Gonzalez-Tallada FJ, Matias-Guiu X et al. DcR1 expression in endometrial carcinomas. Virchows Arch 2010;456:39-44
- 33. Gottwald L, Pasz-Walczak G, Piekarski J et al. Membrane expression of TRAIL receptors DcR1 and DcR2 in the normal endometrium, endometrial atypical hyperplasia and endometrioid endometrial cancer. J Obstet Gynaecol 2014;34:346-9
- 34. Sadarangani A, Kato S, Espinoza N et al. TRAIL mediates apoptosis in cancerous but not normal primary cultured cells of the human reproductive tract. Apoptosis 2007;12:73-85
- 35. Polanski R, Vincent J, Polanska UM et al. Caspase-8 activation by TRAIL monotherapy predicts responses to IAPi and TRAIL combination treatment in breast cancer cell lines. Cell Death Dis 2015;doi:10.1038/cddis.2015.234
- 36. Brincks EL, Kucaba TA, James BR et al. Triptolide enhances the tumoricidal activity of TRAIL against renal cell carcinoma. FEBS J 2015;doi: 10.1111/febs.13532
- 37. Siegelin MD, Reuss DE, Habel A et al. Quercetin promotes degradation of survivin and thereby enhances death-receptor-mediated apoptosis in glioma cells, Neuro Oncol 2009;11:122-31
- 38. Leong S, Cohen RB, Gustafson DL et al. Mapatumumab, an antibody targeting TRAIL-R1, in combination with paclitaxel and carboplatin in patients with advanced solid malignancies: results of a phase I and pharmacokinetic study. J Clin Oncol 2009;27:4413-21
- 39. Jane EP, Premkumar DR, Pollack IF. Bortezomib sensitizes malignant human glioma cells to TRAIL, mediated by inhibition of the NF-{kappa}B signaling pathway. Mol Cancer Ther 2011;10:198-208
- 40. Straughn JM Jr, Oliver PG, Zhou T et al. Anti-tumor activity of TRA-8-anti-death receptor 5 (DR5) monoclonal antibody in combination with chemotherapy and radiation therapy in a cervical cancer model. Gynecol Oncol 2006;101:46-54
- 41. Moxley KM, Chengedza S, Mangiaracina D. Induction of death receptor ligand-mediated apoptosis in epithelial ovarian carcinoma: The search for sensitizing agents. Gynecol Oncol 2009;115:438-42
- 42. Maduro JH, de Vries EG, Meersma GJ et al. Targeting pro-apoptotic TRAIL receptors sensitizes HeLa cervical cancer cells to irradiation-induced apoptosis. Int J Radiation Oncology Biol Phys 2008;72:543-52
- 43. Marini P, Schmid A, Jendrossek V et al. Irradiation specifically sensitises solid tumour cell lines to TRAIL mediated apoptosis. BMC Cancer 2005;5:5
- 44. Jo M, Kim TH, Seol DW et al. Apoptosis induced in normal human hepatocytes by tumor necrosis factor-related apoptosis-inducing ligand. Nat Med 2000;6:564-7
- 45. Volkmann X, Fischer U, Bahr MJ et al. Increased hepatotoxicity of tumor necrosis factor-related apoptosis-inducing ligand in diseased human liver. Hepatology 2007;46:1498-508
- 46. Ganten TM, Sykora J, Koschny R et al. Prognostic significance of tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) receptor expression in patients with breast cancer. J Mol Med 2009;87:995-1007
- 47. Li Y, Jin X, Li J et al. Expression of TRAIL, DR4, and DR5 in bladder cancer: correlation with response to adjuvant therapy and implications of prognosis. Urology 2012;79:968.e7-15
- 48. Younes M, Georgakis GV, Rahmani M et al. Functional expression of TRAIL receptors TRAIL-R1 and TRAIL-R2 in esophageal adenocarcinoma. Eur J Cancer 2006;42:542-7
- 49. Yoldas B, Ozer C, Ozen O et al. Clinical significance of TRAIL and TRAIL receptors in patients with head and neck cancer. Head Neck 2011;33:1278-84
- 50. Ozawa F, Friess H, Kleeff H et al. Effects and expression of TRAIL and its apoptosis-promoting receptors in human pancreatic cancer. Cancer Lett 2001;163:71-81
- 51. Sträter J, Walczak H, Pukrop T et al. TRAIL and its receptors in the colonic epithelium: a putative role in the defense of viral infection. Gastroenterol 2002;122:659-66
- 52. Tomek S, Horak P, Pribill I et al. Resistance to TRAIL-induced apoptosis in ovarian cancer cell lines in overcome by co-treatment with cytotoxic drugs. Gynecol Oncol 2004;94:107-14
- 53. Davidovich IA, Levenson AS, Levenson (Chernokhvostov) VV. Overexpression of DcR1 and survivin in genetically modified cells with pleiotropic drug resistance. Cancer Lett 2004;211:189-97
- 54. Koksal IT, Sanlioglu AD, Karacay B et al. Tumor necrosis factor-related apoptosis inducing ligand-R4 decoy receptor expression is correlated with high Gleason scores, prostate-specific antigen recurrence, and decreased survival in patients with prostate carcinoma. Urol Oncol 2008;26:158-65
- 55. Zhuang L, Lee CS, Scolyer RA et al. Progression in melanoma is associated with decreased expression of death receptors for tumor necrosis factor-related apoptosis-inducing ligand. Hum Pathol 2006;37:1286-94
Następny artykuł:

 Dodaj do ulubionych
Dodaj do ulubionych
