Zespół Turnera – postępowanie diagnostyczno-terapeutyczne
prof. dr hab. n. med. Aneta Gawlik

prof. dr hab. n. med. Aneta Gawlik
- Obraz kliniczny zespołu Turnera – korelacja pomiędzy kariotypem a fenotypem
- Strategia leczenia niedoboru wzrostu będącego głównym objawem zespołu Turnera
- Opieka nad pacjentkami z zespołem Turnera
Zespół Turnera (ZT) zaliczany jest do chorób rzadkich. Częstość jego występowania szacuje się na 25-50 przypadków/100 000 żywo urodzonych noworodków płci żeńskiej, co biorąc pod uwagę liczbę urodzeń w Polsce, przekłada się na 80-100 nowych rozpoznań rocznie (ICD-10: Q96). ZT jest konsekwencją całkowitej lub częściowej monosomii chromosomu X obecnej we wszystkich bądź tylko w niektórych liniach komórkowych. Diagnozę ZT można postawić jedynie u dziewczynki/kobiety z żeńskim fenotypem (żeńskimi narządami płciowymi), u której jednocześnie stwierdza się cechy zespołu (tab. 1) współistniejące z charakterystycznym kariotypem w badaniu cytogenetycznym1 (tab. 2).
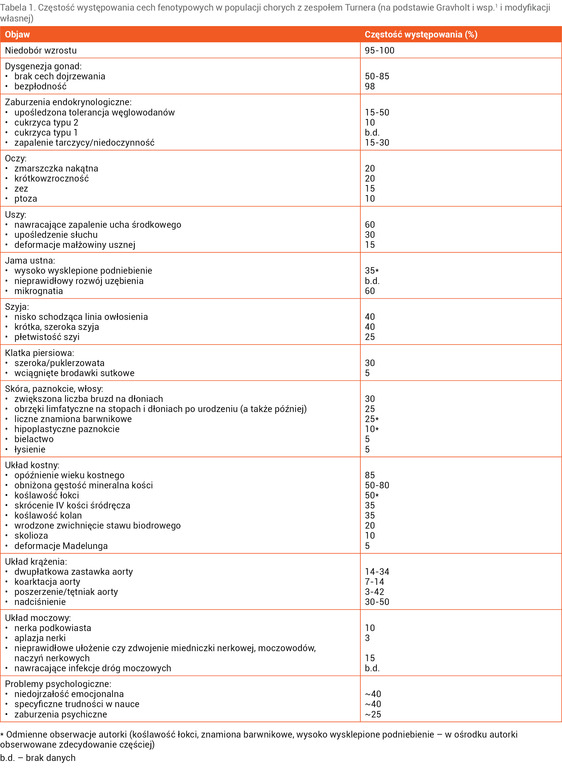
Tabela 1. Częstość występowania cech fenotypowych w populacji chorych z zespołem Turnera (na podstawie Gravholt i wsp.1 i modyfikacji własnej)
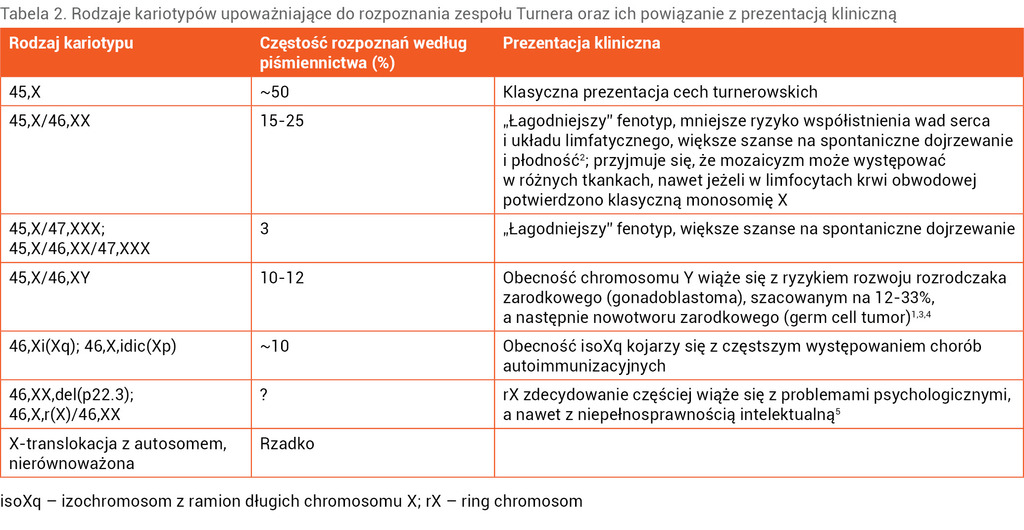
Tabela 2. Rodzaje kariotypów upoważniające do rozpoznania zespołu Turnera oraz ich powiązanie z prezentacją kliniczną
Badanie cytogenetyczne potwierdzające rozpoznanie ZT
Zgodnie z zaleceniami American College of Medical Genetics badanie kariotypu powinno zostać wykonane na podstawie analizy co najmniej 20 komórek6. W przypadku rozpoznań/podejrzeń prenatalnych kariotyp należy ocenić po urodzeniu dziecka. Z uwagi na znaczny postęp w technikach badania sugeruje się również powtórzenie oznaczenia kariotypu u tych pacjentek, u których badanie to wykonano wiele lat temu1.
Nie każda częściowa utrata materiału genetycznego jednego z chromosomów X pozwala na rozpoznanie ZT. W przypadku delecji niewielkiej części ramienia długiego dystalnie do Xq24 możemy spodziewać się izolowanego pierwotnego lub wtórnego braku miesiączki, co nie daje podstawy do rozpoznania ZT. U około 50% pacjentek z ZT w klasycznym badaniu cytogenetycznym z limfocytów krwi obwodowej potwierdza się monosomię 45,X. U prawie 1/3 stwierdza się kariotyp mozaikowy z obecną prawidłową linią 46,XX. U pozostałych występują inne anomalie strukturalne1 chromosomu X (tab. 2).
Prawidłowy wynik badania cytogenetycznego z limfocytów krwi obwodowej u pacjentki z cechami turnerowskimi powinien skłaniać do poszerzenia diagnostyki o kariotyp z fibroblastów skóry1.
Choć nie potwierdzono ścisłej korelacji genotyp–fenotyp, to niektóre rodzaje kariotypów mogą sugerować specyficzny przebieg kliniczny ZT i większe ryzyko chorób współistniejących (tab. 2).
Fenotyp pacjentki z ZT
Rozpoznanie ZT może zostać ustalone przez różnych specjalistów, co wynika z wielu współistniejących problemów zdrowotnych, oraz na różnym etapie życia pacjentki, co z kolei jest związane z różnym nasileniem fenotypu (tab. 1). Wystąpienie cech fenotypowych (tzw. stygmatów), które składają się na zespół objawów ZT, tłumaczy się haploinsuficjencją genu/genów (obecnością tylko jednej prawidłowej kopii genu/genów) zlokalizowanych na chromosomie X7. W prenatalnym badaniu ultrasonograficznym podejrzenie ZT budzą: obrzęk płodu, wodniak torbielowaty, wada układu krążenia czy układu moczowego. Nieprawidłowe stężenia w surowicy α-fetoproteiny, gonadotropiny kosmówkowej, inhibiny A czy wolnego estriolu (test potrójny, test poczwórny) również mogą nasuwać podejrzenie ZT1. Po urodzeniu się dziecka uwagę zwracają: mała urodzeniowa masa i długość ciała, obrzęki limfatyczne stóp i dłoni, płetwistość szyi, deformacja małżowin usznych oraz ich niskie osadzenie, wysoko wysklepione podniebienie oraz zmarszczka nakątna.
W przypadku obecności opisanych cech fenotypowych u noworodka płci żeńskiej konieczne jest wykluczenie typowych wad układu krążenia, w tym przede wszystkim dwupłatkowej zastawki aorty (BAV – bicuspid aortic valve) oraz koarktacji aorty (CoA – coarctation of the aorta), występujących odpowiednio u 16% i 11% pacjentek z ZT. Zasady postępowania w przypadku stwierdzenia wad układu sercowo-naczyniowego zostały bardzo precyzyjnie przedstawione w ostatnio opublikowanych zaleceniach. Poza klasyczną konsultacją kardiologiczną z badaniem elektrokardiograficznym oraz echokardiograficznym, monitorowaniem ciśnienia tętniczego konieczne jest wykonanie rezonansu magnetycznego (MR) w celu pomiaru aorty i oszacowania ryzyka wystąpienia tętniaka rozwarstwiającego aorty. Jest to szczególnie istotne w przypadku młodych kobiet z ZT, u których pęknięcie tętniaka jest najczęstszą przyczyną nagłego zgonu (ryzyko pęknięcia tętniaka znacznie wzrasta w okresie ciąży – spontanicznej lub po zastosowaniu technik wspomaganego rozrodu). Problem nadciśnienia tętniczego narasta z wiekiem (u blisko 30% dziewcząt i 50% dorosłych z ZT) i podobnie jak w przypadku CoA oraz BAV istotnie zwiększa ryzyko rozwarstwienia aorty. Współistniejące wady układu moczowego (nerka podkowiasta czy zdwojenie układu kielichowo-miedniczkowego) mogą przyczyniać się do nawracających zakażeń w drogach moczowych oraz wystąpienia lub nasilenia nadciśnienia tętniczego.
Od okresu przedszkolnego u chorych z ZT charakterystyczne jest zwolnienie tempa wzrastania i niedobór wzrostu. U nastolatek uwagę zwracają brak cech dojrzewania płciowego lub opóźnione dojrzewanie czy zaburzenia miesiączkowania, często pod postacią pierwotnego lub wtórnego braku miesiączki2. ZT predysponuje do dysgenezji gonad i jest klasycznym przykładem hipogonadyzmu hipergonadotropowego. Ponad 90% dziewcząt i kobiet wymaga hormonalnej terapii zastępczej. Gonadotropiny monitoruje się od 10-11 roku życia. Ich wysoka wartość (stężenie hormonu folikulotropowego (FSH – follicle-stimulating hormone) >10 j.m./ml) przy braku rozwoju gruczołów piersiowych może wskazywać na potrzebę indukcji dojrzewania (11-12 rok życia) z zastosowaniem bardzo małych dawek naturalnych estrogenów. W celu zoptymalizowania terapii proponuje się estradiol w formie transdermalnej lub doustnej w dawkach początkowych stanowiących 1/8-1/10 dawki docelowej, do której dochodzi się stopniowo przez 2-3 lata. Dołączenie w odpowiednim momencie progestagenów jest konieczne, aby zapobiec przerostom endometrium1,8.
ZT często kojarzy się ze zwiększonym ryzykiem wystąpienia chorób autoimmunizacyjnych, stąd zarówno w chwili rozpoznania zespołu, jak i w trakcie obserwacji konieczne jest wykonanie badań i monitorowanie w kierunku: choroby Hashimoto (autoimmunizacyjnego zapalenia tarczycy), celiakii, cukrzycy, bielactwa, łysienia plackowatego czy nieswoistego zapalenia jelit9,10. Częstym i narastającym z wiekiem problemem są zaburzenia słuchu, w dzieciństwie występujące głównie wskutek nawracających zapaleń ucha środkowego (niedosłuch przewodzeniowy), natomiast w życiu dorosłym mające charakter niedosłuchu odbiorczego.
Diagnoza ZT zobowiązuje do przeprowadzania u chorych regularnych badań kontrolnych, w dużej mierze wykonywanych cyklicznie jako badania profilaktyczne, oraz w przypadku potwierdzenia chorób współistniejących wymaga objęcia pacjentek stałą opieką wielospecjalistyczną (tab. 3).
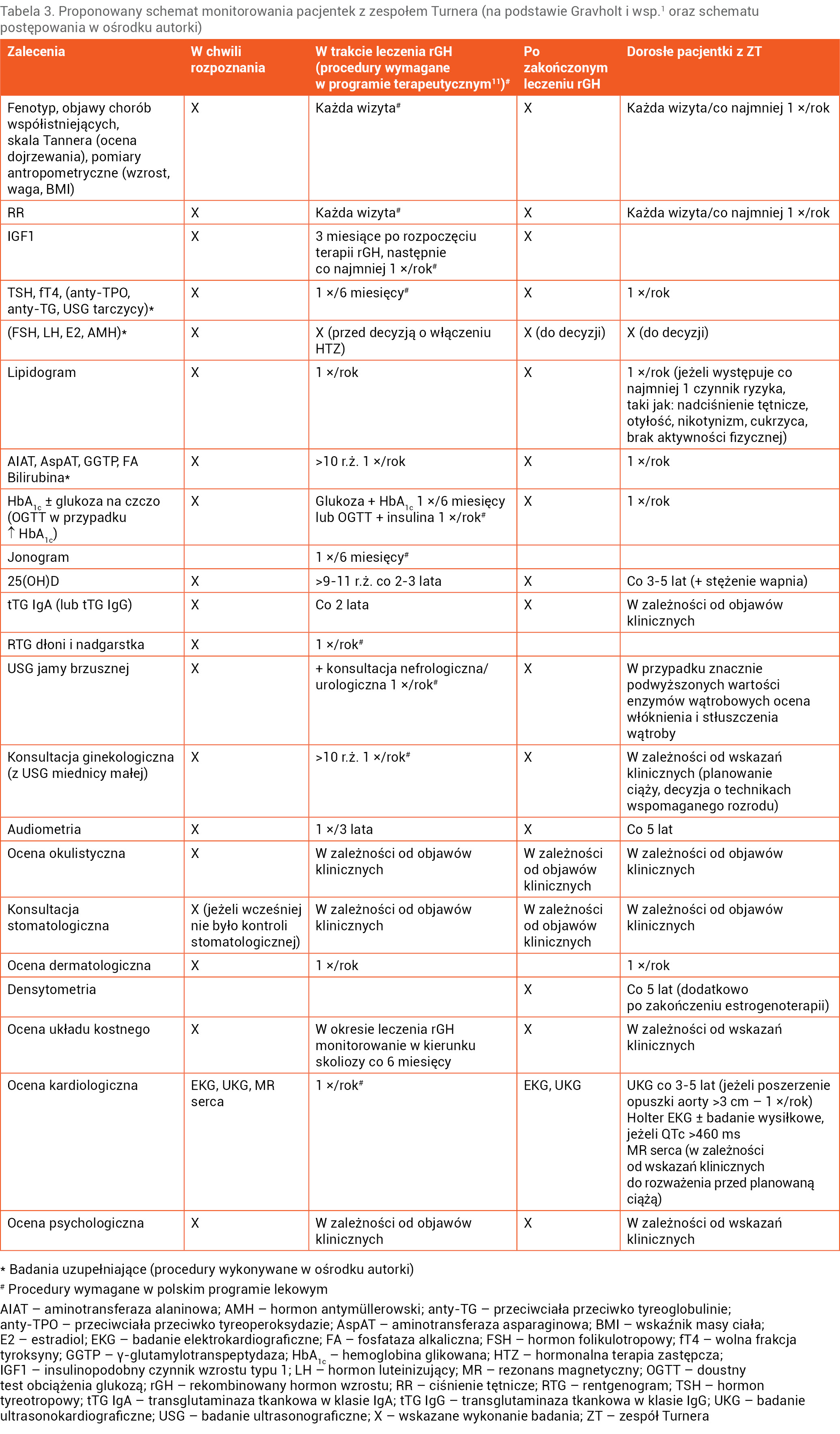
Tabela 3. Proponowany schemat monitorowania pacjentek z zespołem Turnera (na podstawie Gravholt i wsp.1 oraz schematu postępowania w ośrodku autorki)