Stopień nasilenia dysmorfii może być różny i niekoniecznie wpływa na szybkość postawienia diagnozy. Średni wiek rozpoznania ZT w populacji polskiej szacuje się na 9-10 rok życia, czyli 2 lata później niż w populacji amerykańskiej12,13. Najczęstszymi powodami zlecenia wykonania badania cytogenetycznego są niedobór wzrostu stwierdzany u prawie wszystkich pacjentek oraz objawy związane z niewydolnością gonad obserwowane w różnym nasileniu u około 90% zdiagnozowanych.
Spontaniczne wzrastanie w ZT
Niedobór wzrostu jest głównym objawem ZT i dotyczy 95-100% badanych1. Ich spontaniczne wzrastanie w ZT ma charakterystyczny przebieg i w ciągu życia wyróżnia się kilka jego faz:
- okres wewnątrzmacicznego zahamowania wzrastania (IUGR – intrauterine growth restriction)
- okres od urodzenia do 3 roku życia z prawidłowym tempem wzrastania i spowolnioną progresją wieku kostnego w stosunku do wieku metrykalnego
- okres od 3 do 12 roku życia z opóźnionym tempem wzrastania i prawidłową progresją wieku kostnego
- okres po 12 roku życia z opóźnionym tempem wzrastania i spowolnioną progresją wieku kostnego.
Konsekwencją opóźnienia wieku kostnego jest wydłużenie okresu wzrastania nawet do początku trzeciej dekady życia. Nie wiąże się to jednak z poprawą prognozy wzrostu ostatecznego. Wzrost końcowy nieleczonych pacjentek jest przeciętnie o 18-20 cm niższy od prognozowanego (ryc. 1). W celu wiarygodnej oceny spontanicznego tempa wzrastania zaleca się używanie siatek centylowych dla ZT, a w przypadku stwierdzenia odchylania się od dotychczasowego kanału centylowego ważne jest poszukiwanie dodatkowej przyczyny nieprawidłowego wzrastania (np. niedoczynności tarczycy w przebiegu przewlekłego zapalenia, celiakii czy nieswoistego zapalenia jelit).
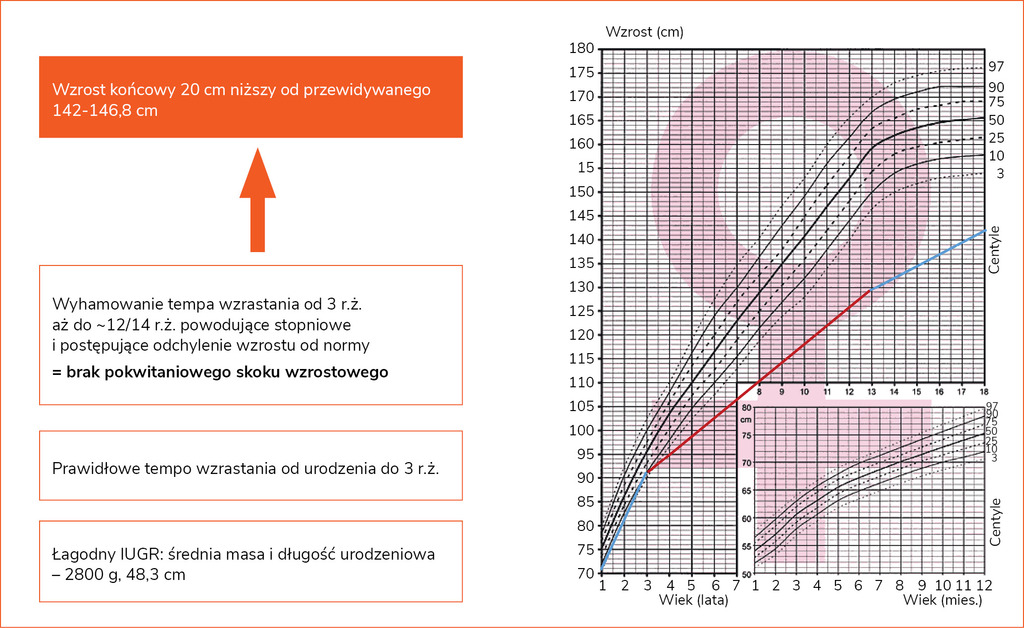
Rycina 1. Przykładowy spontaniczny przebieg tempa wzrastania u nieleczonej pacjentki z zespołem Turnera
Niskorosłość w ZT nie wynika z niedoboru hormonu wzrostu. Za zwolnienie tempa wzrastania oraz niski wzrost w dużej mierze odpowiada haploidalność genu SHOX (short stature homeobox-containing gene) zlokalizowanego w pseudoautosomalnym regionie ramienia krótkiego chromosomów płciowych. Gen ten koduje czynnik transkrypcyjny, kluczowy dla prawidłowego rozwoju układu kostnego, w szczególności kości kończyn.
Terapia hormonalna poprawiająca wzrost końcowy
W Polsce ZT jest jednym ze wskazań do refundacji leczenia hormonem wzrostu. Historia stosowania hormonu wzrostu w terapii niedoboru wzrostu sięga lat 60. XX wieku, a jego skuteczność w populacji z ZT potwierdzono w opublikowanym w 2005 roku badaniu kanadyjskim (ryc. 2), które było badaniem randomizowanym kontynuowanym do osiągnięcia niemalże wzrostu ostatecznego14. Pacjentki były leczone od 5,5-7,6 roku życia i w porównaniu z obserwowaną w tym czasie grupą kontrolną ich wzrost końcowy poprawił się o 5-8 cm. Podobne efekty obserwowano też w innych badaniach15.

Rycina 2. Rys historyczny wykorzystania hormonu wzrostu w terapii niskorosłości u pacjentek z zespołem Turnera
Uwzględniając profil spontanicznego wzrastania (ryc. 1), leczenie rekombinowanym hormonem wzrostu (rGH – recombinant growth hormone) dziewcząt z ZT optymalnie należałoby rozpoczynać w 4-6 roku życia, aby uzyskać wzrost w granicach normy wiekowej i uniknąć dużych różnic w rozwoju fizycznym w porównaniu z rówieśniczkami. Oczywiście czas rozpoczęcia terapii w dużej mierze zależy od wieku, w którym postawiono diagnozę. Dla uzyskania oczekiwanych korzyści z leczenia powinno ono zostać zainicjowane przed 12-13 rokiem życia. W polskich warunkach refundacja leczenia w ramach programu terapeutycznego przysługuje tylko w przypadku wzrostu poniżej 3 pc (percentyla) według siatek centylowych dla zdrowej populacji żeńskiej. Międzynarodowe towarzystwa rekomendują rozpoczęcie terapii także w przypadku zwolnienia tempa wzrastania (<50 pc na podstawie 6-miesięcznej obserwacji), jak również w przypadku niekorzystnej prognozy wzrostowej wynikającej np. z niskiego wzrostu rodziców czy zaawansowanego dojrzewania (dojrzewające spontanicznie dziewczęta z ZT mają bardziej zaawansowany wiek kostny, co skraca okres wzrastania i ostatecznie skazuje na niski wzrost końcowy)1.
Leczenie rGH rozpoczyna się od dawki 45-50 µg/kg/24 h (odpowiednik około 1 j.m./kg/tydzień lub 1,3-1,5 mg/m2/ 24 h bądź 4-4,5 j.m./m2/24 h) stosowanej w codziennych wieczornych podskórnych iniekcjach. Szkolenie rodziców/opiekunów oraz pacjentek w zakresie obsługi penów do podawania leku w warunkach domowych odbywa się w ośrodkach prowadzących programy terapeutyczne. Dawka rGH jest systematycznie modyfikowana przez lekarza prowadzącego podczas wizyt kontrolnych odbywających się najczęściej co 3-6 miesięcy. Poza masą/powierzchnią ciała bierze się pod uwagę efekty terapeutyczne (tempo wzrastania) oraz stężenie insulinopodobnego czynnika typu 1 (IGF1 – insulin-like growth factor type 1). W większości krajów kryterium zakończenia terapii rGH jest wiek kostny 14 lat oraz wyhamowanie wzrastania poniżej 2-3 cm w skali roku, a według standardów polskich również osiągnięcie wysokości 158 cm (10 pc według siatek dla zdrowej populacji).
Czynnikami predykcji wyższego wzrostu końcowego po zakończeniu terapii rGH są:
- wyższy wzrost w chwili inicjacji leczenia
- wyższy wzrost rodziców (większa średnia wysokość ciała rodziców [MPH – mid-parental height])
- młodszy wiek rozpoczęcia leczenia
- dłuższy czas stosowania rGH przed rozpoczęciem dojrzewania
- dłuższy czas leczenia i większa dawka rGH1.
Za wiarygodny wskaźnik odpowiedzi na leczenie przyjmuje się tempo wzrastania w pierwszych 2 latach terapii. Znaczenie mają też kontrolowanie i leczenie chorób towarzyszących – w każdym przypadku niewystarczającej odpowiedzi na rGH należy wykluczyć współistnienie chorób przewlekłych (np. zaburzeń wchłaniania w przebiegu celiakii, niedoboru witaminy D3, niedoczynności tarczycy).
Przed rozpoczęciem terapii należy poinformować o potencjalnie mogących wystąpić działaniach niepożądanych. Włączenie leczenia nakłada obowiązek ścisłego monitorowania gospodarki węglowodanowej, zwrócenia uwagi na objawy wzmożonego ciśnienia śródczaszkowego oraz złuszczenia głowy kości udowej16, obserwacji w kierunku rozwoju skoliozy bądź pogłębiania się już istniejącej17, objawów zapalenia trzustki1. W trakcie leczenia należy unikać zbyt dużych stężeń IGF1 (oznaczenia kontrolne wykonywane są przynajmniej raz w roku), tj. powyżej 2 odchyleń standardowych (SD – standard deviation). W przypadku podwyższonych stężeń IGF1 wskazana jest redukcja dawki rGH. Dane z dużego rejestru nie potwierdziły związku stosowania rGH ze zwiększonym ryzykiem rozrostu nowotworowego w populacji ZT17-19.
W przypadku znacznego deficytu wzrostowego i słabej prognozy wzrostu końcowego u dziewcząt powyżej 10 roku życia można rozważyć dołączenie steroidu anabolicznego (oksandrolonu; w Polsce sprowadzany w ramach importu docelowego). Większe dawki tego leku mogą niekorzystnie wpływać na rozwój gruczołów piersiowych, być przyczyną objawów wirylizacji (powiększenie łechtaczki, niski głos, trądzik, hirsutyzm) oraz przyspieszenia wieku kostnego, dlatego rekomenduje się podawanie niższych dawek – 0,03 mg/kg/24 h. W trakcie leczenia wskazane jest monitorowanie enzymów wątrobowych. Korzyść wynikająca z zastosowania tego leku w terapii skojarzonej z rGH to wzrost końcowy wyższy o 2-5 cm w porównaniu z osobami leczonymi jedynie rGH1,20,21. Dołączanie bardzo małych dawek estrogenów do leczenia rGH przed okresem fizjologicznego dojrzewania pacjentki w celu poprawy wzrostu nie jest aktualnie rekomendowane i wymaga dalszych badań1.