III Kongres Akademii po Dyplomie Okulistyka już w ten piątek! Kup bilet i dołącz do ekspertów podczas Siatkówka Meeting! Sprawdź >
Zaburzenia krzepnięcia w chirurgii
Wybrane osoczowe mechanizmy powstawania koagulopatii pokrwotocznej
Janusz Trzebickia,b, Piotr Paluszkiewicza,c,d, Ewa Mayzner-Zawadzkaa,e
Chirurgia po Dyplomie 2013; 8 (1): 42-46
Streszczenie
Postęp w badaniach nad kontrolą hemostazy ustroju oraz istotne kliniczne implikacje terapeutyczne w odniesieniu do układu krzepnięcia krwi i fibrynolizy wymagają okresowego uaktualniania wytycznych postępowania lekarskiego u chorych z masywną utratą krwi. W niniejszym artykule skupiono się na aspektach klinicznych końcowego etapu formowania skrzepu i aktywności fibrynolitycznej osocza u chorych z masywną utratą krwi. Omówiono mechanizm wczesnej aktywacji białka C, powstawanie nadmiernej fibrynolizy pokrwotocznej i pourazowej oraz opisano terapię preparatami fibrynogenu u chorych z masywną utratą krwi. Odniesiono się do najnowszych informacji sugerujących farmakologiczne zahamowanie fibrynolizy w okresie okołooperacyjnym i pourazowym. Przedstawione informacje mogą stanowić asumpt do uaktualnienia zaleceń postępowania terapeutycznego z chorymi, u których nastąpiła masywna utrata krwi.
Wprowadzenie
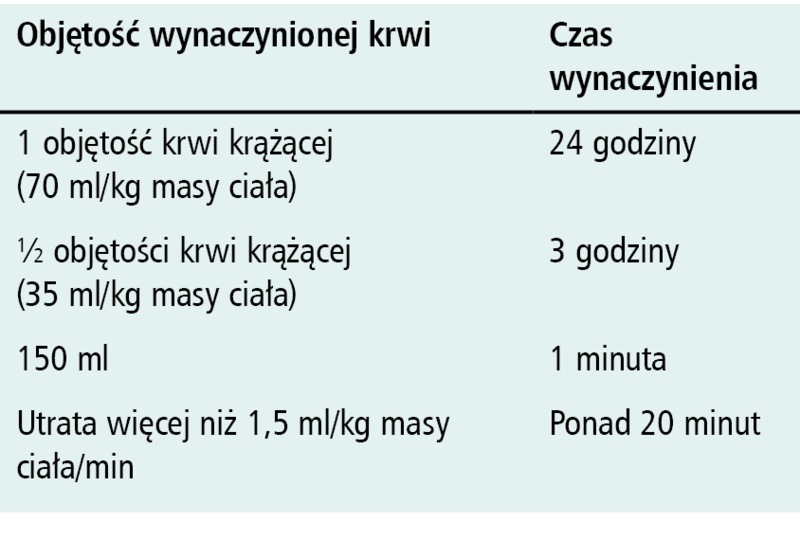
Tabela. Definicje masywnej utraty krwi wg Stainsby’ego [2] uwzględniające utraconą objętość krwi krążącej i dynamikę utraty
Utrata krwi jest zjawiskiem nieodłącznie towarzyszącym dyscyplinom zabiegowym. Z klinicznego punktu widzenia krwawienie jest stanem naglącym i zawsze powinno być traktowane jako stan zagrożenia życia. Masywnym krwawieniem określa się zwykle utratę 20% lub więcej całkowitej objętości krwi krążącej [1]. Bardziej precyzyjne określenie masywnej utraty krwi, uwzględniające dynamikę krwotoku, przedstawili Stansby i wsp. [2] (tabela). Niezależnie od zastosowanych kryteriów, masywnej utracie krwi towarzyszą różne następstwa patofizjologiczne doprowadzające do zaburzeń ogólnoustrojowych w następstwie hipoperfuzji tkankowej. Zmniejszenie perfuzji tkanek uniemożliwiające utrzymanie metabolizmu tlenowego uznawane jest obecnie za podstawę patofizjologiczną klinicznie definiowanego wstrząsu. Wystąpienie takiej sytuacji klinicznej w następstwie utraty krwi określane jest mianem wstrząsu krwotocznego [3].
Zaburzenia krzepnięcia krwi u chorych krwawiących są istotną klinicznie odpowiedzią układu hemostazy na wstrząs. Rozwijające się zaburzenia hemostazy uznawane są za najczęstszą przyczynę wykrwawienia we wczesnym okresie po urazie [4]. Udowodniono, że objętość przetaczanej krwi w okresie pourazowym lub pooperacyjnym jest jednym z najważniejszych czynników rokowniczych przeżycia i częstości wystąpienia powikłań [5].
Obecnie przyjmuje się, że zapobieganie rozwojowi zaburzeń krzepnięcia krwi może być kluczowym celem terapeutycznym u chorych masywnie krwawiących i przyczynić się do poprawy wyników leczenia. Z chirurgicznego punktu widzenia za najważniejszy element leczenia należy uznać dążenie do powstrzymania krwotoku. Ustalenie takiego priorytetu wymaga przyjęcia szczególnej taktyki postępowania polegającej na stosowaniu uproszczonych technik operacyjnych ograniczających utratę krwi (hemostatic surgery) z następową resuscytacją, wyrównaniem hipotermii i kwasicy oraz wtórnym zabiegami operacyjnymi (stage surgery) [6]. Postępowanie zgodne z tymi zasadami znacznie poprawiło wyniki leczenia chorych z ciężkimi obrażeniami ciała [7-10].
Badania nad mechanizmami powstawania zaburzeń krzepnięcia krwi u chorych masywnie krwawiących umożliwiły częściowe poznanie możliwości zapobiegania koagulopatii pourazowej i pokrwotocznej. Wysoki stopień skomplikowania występujących zaburzeń stwarza konieczność prowadzenia terapii wielokierunkowej i multidyscyplinarnej. Dodatkowym zjawiskiem jest fizjologiczna odpowiedź ustroju na niewystarczającą perfuzję tkanek, polegająca na uruchomieniu mechanizmów zapobiegających krzepnięciu przez hamowanie kaskadowej aktywacji osoczowych czynników krzepnięcia i aktywacji fibrynolizy [11].
Konieczność uporządkowania informacji przedstawianych w piśmiennictwie dała asumpt do stworzenia ogólnych zaleceń postępowania u chorych, u których doszło do masywnej utraty krwi z różnych powodów. W ostatnich latach opisano podstawowe cele terapeutyczne w formie rekomendacji ekspertów europejskich [6], aktualizowane w miarę postępu prac nad mechanizmami rozwoju zaburzeń krzepnięcia krwi u ofiar ciężkich wypadków [12]. W warunkach polskich przeprowadzono badanie, którego celem było uzyskanie uśrednionej opinii polskich ekspertów dotyczącej zapobiegania i rozpoznawania koagulopatii pokrwotocznej oraz metod wspomagających leczenie masywnej utraty krwi. Wyniki tego badania przedstawiono w formie polskich zaleceń pod auspicjami Stowarzyszenia na Rzecz Leczenia Ciężkich Krwotoków [13]. Obecnie należy przyjąć, że przedstawiane zalecenia europejskie i krajowe częściowo utraciły swą aktualność z powodu ciągłego dostarczania nowych informacji o możliwościach rozpoznawania i leczenia nabytych zaburzeń krzepnięcia krwi.
Mechanizm aktywacji białka C u chorych z niewystarczającą perfuzją tkanek
Poszukiwanie przyczyn zaburzeń krzepnięcia krwi u chorych we wstrząsie umożliwiło postawienie hipotezy o kluczowej roli aktywacji lub uszkodzenia śródbłonka naczyniowego. Dotychczas nie przedstawiono dokładnego mechanizmu aktywacji śródbłonka, przypuszcza się jednak, że zasadniczą rolę odgrywa zmiana architektoniki glikokaliksu spowodowana zmniejszeniem dynamiki przepływu krwi w mikrokrążeniu [14]. Obecnie nie można wykluczyć autoheparynizacji w następstwie uszkodzenia glikokaliksu i uwolnienia wielocukrów o działaniu zbliżonym do heparyny [15]. Skutkiem aktywacji śródbłonka naczyń jest również nadekspresja trombomoduliny wiążącej powstającą w miejscu uszkodzenia naczyń trombinę, następnie aktywacja białka C i zahamowanie kaskady krzepnięcia krwi. U chorych z rozwiniętymi zaburzeniami krzepnięcia krwi w okresie pourazowym stwierdzono zwiększone stężenie trombomoduliny. Stężenie trombomoduliny korelowało też ze śmiertelnością po urazach [11]. Zwiększeniu stężenia trombomoduliny towarzyszyło zmniejszenie stężenia nieaktywnej formy białka C. Niezależnie od innych czynników, zjawisko to wiązało się ze zwiększeniem śmiertelności chorych bezpośrednio po urazie [11].
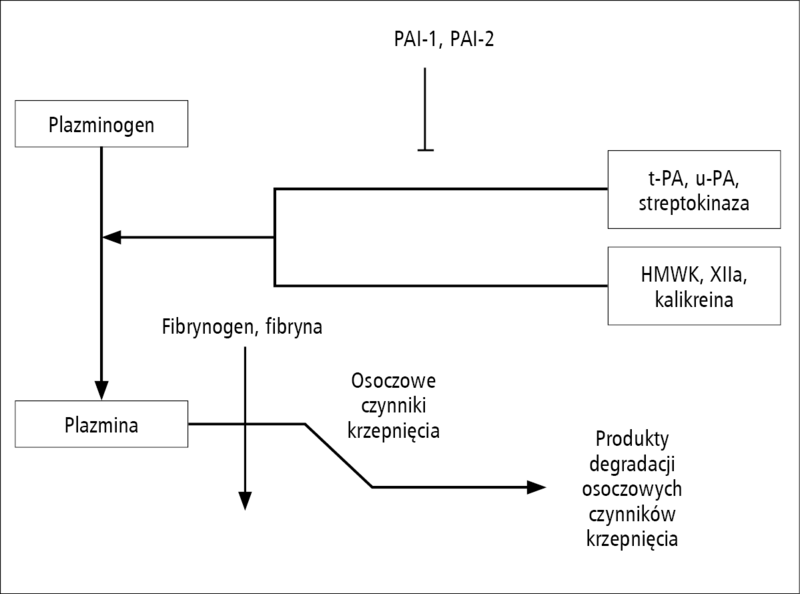
Rycina 1. Podstawowe mechanizmy fibrynolizy z uwzględnieniem niektórych aktywatorów i inhibitorów aktywacji plazminy. PAI-1 i PAI-2 – inhibitor aktywatora plazminogenu typu 1 i 2, t-PA i u-PA – tkankowe aktywatory plazminy, odpowiednio tkankowy i urokinaza, HMWK – wysokocząsteczkowy kininogen, FDP – produkty degradacji fibrynogenu.
Nadmierna fibrynoliza
Sprawność układu fibrynolitycznego umożliwia fizjologiczną kontrolę ustroju nad rozmiarem tworzących się skrzeplin oraz odpowiada za przywrócenie przepływu w uszkodzonym naczyniu (ryc. 1). Wielokrotnie udowodniono znaczenie nadmiernej fibrynolizy w rozwoju pourazowych zaburzeń krzepnięcia krwi [16]. Trwają liczne badania mające na celu poznanie szczegółowych mechanizmów odpowiedzialnych za pobudzenie układu fibrynolitycznego [17]. Interesujące są dowody na udział plazminy i jej aktywatorów w regulacji aktywności metaloproteinaz tkankowych (matrix metalloproteinase, MMP), ich inhibitorów (tissue inhibitor of metalloproteinase, TIMP) oraz receptorów lipoprotein o małej gęstości (lipoprotein receptor, LPR) odpowiadających za funkcję bariery krew-mózg [18,19]. Stwierdzono, że nadmierna fibrynoliza jest ważnym następstwem wstrząsu [20], koagulopatii położniczej [21], ciężkich urazów [22], niewydolności wątroby [23], fazy anhepatycznej przeszczepienia wątroby [24], uszkodzenia ośrodkowego układu nerwowego [25] oraz stosowania krążenia pozaustrojowego [26]. Przypuszcza się, że farmakologiczne ograniczenie fibrynolizy może zmienić częstość występowania jawnych koagulopatii [16], zmniejszyć konieczność przetaczania preparatów krwi oraz poprawić dotychczasowe wyniki leczenia [27]. W badaniu przeprowadzonym z udziałem chorych, którzy doznali ciężkich obrażeń ciała, zastosowanie kwasu traneksamowego pozwoliło na zmniejszenie całkowitej śmiertelności pourazowej o 9% oraz zmniejszenie śmiertelności zależnej od wykrwawienia o 15% w porównaniu z grupą kontrolną otrzymującą placebo [28]. Wykazano też zmniejszenie śmiertelności po leczeniu kwasem traneksamowym chorych z krwawieniem do przewodu pokarmowego. Wyniki metaanalizy [29] wskazują, że stosowanie kwasu traneksamowego zmniejsza śmiertelność o 39% w porównaniu z obserwowaną wśród chorych otrzymujących placebo. W podgrupach chorych leczonych technikami endoskopowymi lub antagonistami receptorów histaminowych typu 2 (cymetydyna) albo inhibitorami pompy protonowej (lanzoprazol) nie stwierdzono korzyści terapeutycznych w porównaniu z obserwowanymi w grupie chorych otrzymujących wyłącznie lek antyfibrynolityczny [29]. Zastosowanie kwasu traneksamowego ogranicza częstość przetaczania krwi u kobiet z masywnymi krwawieniami miesiączkowymi i wykazuje przewagę nad niesteroidowymi lekami przeciwzapalnymi, progestagenami oraz etamsylatem [30]. Nie stwierdzono natomiast wpływu kwasu traneksamowego na zmniejszenie częstości przetaczania krwi u chorych poddawanych przeszczepieniu wątroby [31]. W metaanalizie danych pochodzących od 1913 chorych uczestniczących 33 badaniach wskazano jednak, że rozpoznanie hiperfibrynolizy, jej osłabienie za pomocą aprotyniny lub ograniczenie jej następstw dzięki podaniu rekombinowanego aktywnego czynnika krzepnięcia VII (rFVIIa) prawdopodobnie zmniejsza nasilenie krwawienia i ogranicza zużycie preparatów krwi [31].
Stężenie fibrynogenu
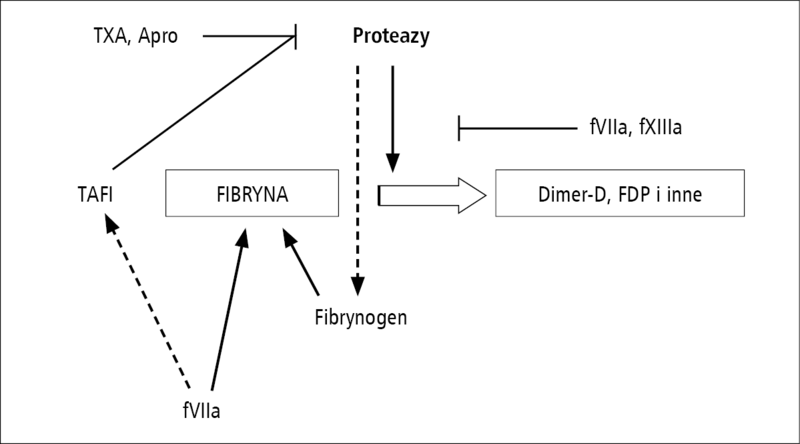
Rycina 2. Wybrane drogi stabilizacji fibryny. TXA – kwas traneksamowy, Apro – aprotynina, fVIIa – aktywny VII czynnik krzepnięcia, fXIIIa – aktywny XIII czynnik krzepnięcia, TAFI – inhibitor fibrynolizy aktywowany przez trombinę, FDP – produkty degradacji fibrynogenu.
Hemostaza wtórna jest procesem ukierunkowanym na wytworzenie nierozpuszczalnej fibryny z fibrynogenu, będącego osoczową glikoproteiną syntetyzowaną w wątrobie (ryc. 2). Stężenie fibrynogenu w osoczu zależy od jego syntezy, zużycia, utraty podczas krwotoków lub rozcieńczenia w następstwie leczenia płynami [32,33]. Stosowanie roztworów koloidowych może dodatkowo wpływać na procesy polimeryzacji fibryny [34,35]. Stężenie fibrynogenu w osoczu jest niezależnym czynnikiem ryzyka śmierci chorych krwawiących [36]. Podczas leczenia chorych z masywną utratą krwi należy dążyć do utrzymania stężenia fibrynogenu powyżej 2 g/l [37,38].
Najczęściej wykorzystywane źródła egzogennego fibrynogenu to świeżo mrożone osocze (fresh frozen plasma, FFP), krioprecypitat (KRIO) i liofilizowane koncentraty fibrynogenu ludzkiego (FIB) [39]. Zawartość fibrynogenu w tych preparatach jest zróżnicowana, co utrudnia prowadzenie precyzyjnej terapii substytucyjnej. Ich przetaczanie stwarza również ryzyko przenoszenia wirusów, a w przypadku FFP wystąpienia ostrego potransfuzyjnego uszkodzenia płuc i hiperwolemii [40]. W przeciwieństwie do FFP i KRIO FIB zawiera wystandaryzowaną ilość fibrynogenu, jest bezpieczniejszy pod względem wirusologicznym i gotowy do użycia w ciągu kilku minut. W ostatnich latach wzrasta wykorzystywanie FIB w nabytych hipofibrynogenemiach pourazowych i pooperacyjnych [41], co pozwala zmniejszyć objętość przetaczanych płynów i ryzyko przenoszenia zakażeń wirusowych.