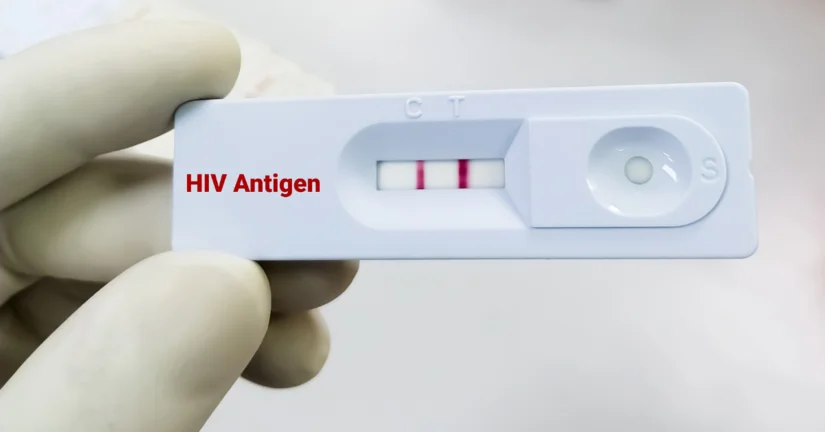
Od 18-25 listopada odbywa się Europejski Tydzień Testowania (ETT) HIV oraz HCV. Inicjatywa zachęca do zwiększonego testowania i promowania wczesnej diagnozy. Kampania odbywa się równolegle w wielu krajach Europy, a jednym z najważniejszych jej celów jest zwiększenie dostępności testów oraz podniesienie świadomości na temat korzyści płynących z wczesnego wykrywania HIV i HCV.
Lekarze mają szansę pomóc w walce z HIV
Europejski Tydzień Testowania to okazja, aby lekarze włączyli się w działania na rzecz profilaktyki HIV i HCV. To właśnie oni w swoich gabinetach mogą zasygnalizować pacjentom, jak ważne jest wykonanie takiego testu – tym bardziej, że w ramach tej akcji, testy na HIV są dostępne bezpłatnie i anonimowo.
- W tym czasie jednoczymy siły z organizacjami społecznymi, przedstawicielami opieki zdrowotnej oraz partnerami w dziedzinie zdrowia publicznego, aby zwiększyć świadomość i dostępność testów. Razem możemy promować wczesną diagnozę i leczenie! – zachęca m.in. Główny Inspektorat Sanitarny.
Testuj. Lecz. Zapobiegaj.
Kampania odbywa się pod hasłem „Testuj. Lecz. Zapobiegaj.” Organizowane są różne działania, a najważniejszym z nich jest dostęp do testów w specjalnych Punktach Konsultacyjno-Diagnostycznych (PKD). To właśnie te miejsca dają pacjentom możliwość wykonania testu na HIV bez potrzeby skierowania, w pełnej anonimowości. W najbliższych dniach niektóre z punktów rozszerzają swoją działalność, oferować będą szybkie testy, dzięki którym pacjent może poznać wynik już po kilku minutach. W niektórych punktach dostępne są także testy na inne choroby przenoszone drogą płciową, jak kiła czy HCV. W Warszawie najdłużej, do g. 20 działa punkt przy ul. Marszałkowskiej 68/70.
Kiedy i gdzie można wykonać test?
Z danych Krajowego Centrum ds. AIDS wynika, że HIV w Polsce wciąż stanowi poważne zagrożenie zdrowotne. Choć testowanie w kierunku HIV stało się bardziej dostępne, to wciąż zbyt wielu pacjentów nie zdaje sobie sprawy z ryzyka, jakie niosą niewykryte zakażenia.
Więcej informacji na temat dostępnych punktów testowania HIV w całej Polsce można znaleźć na stronie Krajowego Centrum ds. AIDS (aids.gov.pl/pkd).
Warto też przypomnieć bezpłatny nr 800 888 448 czynny we wszystkie dni robocze w godzinach 9.00 – 21.00 oraz w dniach 01.11 – 21.12.2024 r. w weekendy od godz. 21.00 w piątki do 09.00 w poniedziałki.