Podejrzenie obecności zmian infiltracyjnych we wtórnych kardiomiopatiach restrykcyjnych
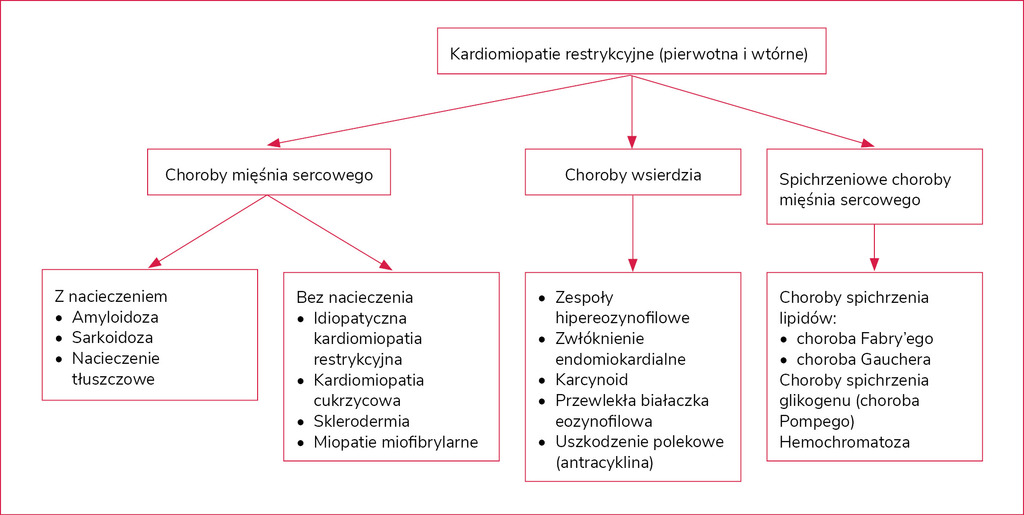
Rycina 5. Podział kliniczny kardiomiopatii restrykcyjnych
Kardiomiopatie restrykcyjne stanowią szeroką grupę schorzeń obejmujących zarówno zmiany w mięśniu sercowym, jak i we wsierdziu (ryc. 5). W grupie tej znajduje się zarówno kardiomiopatia restrykcyjna pierwotna typu mieszanego (idiopatyczna), jak i cała grupa wrodzonych i nabytych kardiomiopatii wtórnych22. Ich dominującą cechą kliniczną jest dysfunkcja rozkurczowa restrykcyjna w badaniu echokardiograficznym przy jednoczesnym zachowaniu funkcji skurczowej. Podejrzenie kardiomiopatii restrykcyjnej z nacieczeniem kwalifikuje chorego do diagnostyki biopsyjnej. Dotyczy to głównie przypadków podejrzenia:
- amyloidozy serca
- sarkoidozy serca.
Amyloidoza serca jest coraz częściej rozpoznawaną przyczyną niejasnej etiologicznie NS. Jest chorobą zarówno nabytą, jak i mającą podłoże genetyczne. Wykazano jak do tej pory 11 różnych białek odkładających się w sercu w formie amyloidu23. Statystycznie najczęściej (80% przypadków) obserwuje się złogi łańcuchów lekkich (amyloidoza AL), a następnie transtyretyny (amyloidoza ATTR, 18% przypadków). W wycinkach mięśnia sercowego transtyretyna wykrywana jest jednak najczęściej24. Potwierdzają to także badania własne autora. Ten typ amyloidozy występuje w formie nabytej („wild type") i wrodzonej, związanej z mutacjami w genie transtyretyny. Ze względu na pogrubienie przegrody międzykomorowej w następstwie odkładania się depozytów amyloidu (głównie w jej części tylno-podstawnej) choroba ta często wykazuje cechy kardiomiopatii przerostowej.
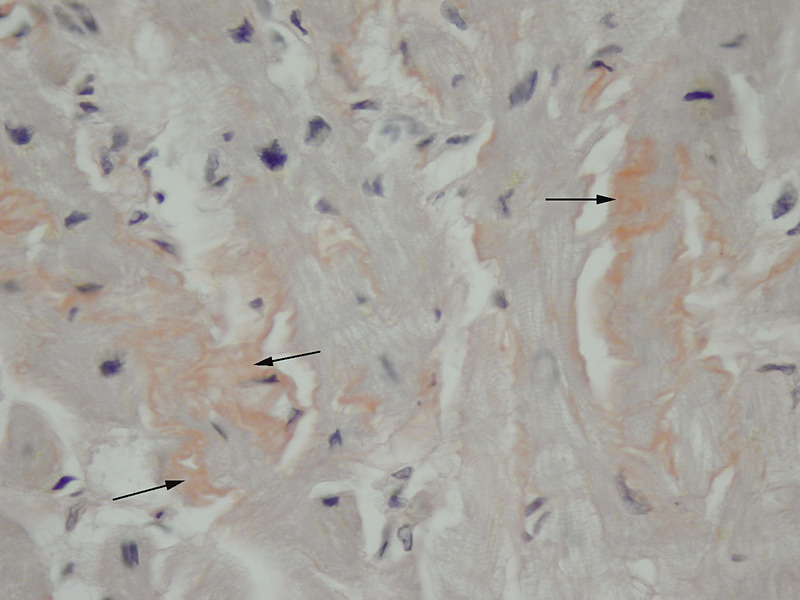
Rycina 6. Dodatnie barwienie histochemiczne czerwienią Kongo wycinków mięśnia sercowego na obecność złogów amyloidu (strzałki)
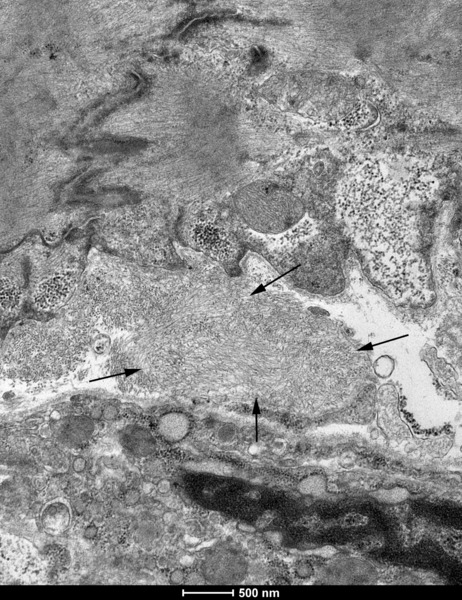
Rycina 7. Obecność włókienek amyloidozy transtyretynowej (strzałki) w wycinkach mięśnia sercowego w badaniu w mikroskopie elektronowym
Najbardziej rozpowszechnioną metodą histochemiczną pozwalającą na potwierdzenie obecności amyloidu jest barwienie histochemiczne czerwienią Kongo (ryc. 6). Techniki immunohistochemiczne umożliwiają natomiast typowanie amyloidu, będące podstawą odpowiedniego postępowania leczniczego. Transmisyjna mikroskopia elektronowa umożliwia wykazanie zagregowanych filamentów amyloidu o długości 7-10 nm (ryc. 7). Planując diagnostykę biopsyjną amyloidozy, należy uwzględnić zabezpieczenie 1 wycinka do badania w mikroskopie elektronowym (utrwalanie w 2,5-3% glutaraldehydzie w buforze kakodylowym/fosforanowym). Algorytm postępowania przy podejrzeniu amyloidozy serca przedstawiono w monografii poświęconej kardiomiopatii restrykcyjnej22.
Sarkoidoza serca jest wielonarządową chorobą o nieustalonej etiologii, cechującą się tworzeniem w narządach ziarniniaków bez towarzyszących zmian martwiczych. Zwykle obserwuje się także obecność komórek olbrzymich. Z tego powodu sarkoidozę różnicuje się z olbrzymiokomórkowym ZMS. Występowanie wysoce zorganizowanych ziarniniaków wraz z towarzyszącym włóknieniem jest cechą patognomoniczną sarkoidozy. Jednak zaledwie u 25% chorych z kardiomiopatią rozstrzeniową i rozpoznaną klinicznie sarkoidozą udaje się w materiale biopsyjnym wykazać ich obecność25. Jest to związane z preferencyjną lokalizacją ziarniniaków w miokardium, następnie epikardium, a dopiero na końcu w endokardium dostępnym biopsyjnie. Klinicznie sarkoidoza serca zwykle przebiega pod postacią NS z towarzyszącymi zaburzeniami rytmu i przewodnictwa26. Z uwagi na dobrą odpowiedź choroby na leczenie immunosupresyjne biopsja endomiokardialna stanowi istotny element jej diagnostyki.
Podejrzenie obecności zmian spichrzeniowych we wtórnej kardiomiopatii restrykcyjnej
Kardiomiopatie restrykcyjne o podłożu spichrzeniowym są kardiomiopatiami uwarunkowanymi genetycznie, w przebiegu których obserwuje się wielonarządowe objawy kliniczne. Mutacje genowe mogą być zarówno jedno- (choroba Fabry’ego, choroba Gauchera), jak i wielowariantowe (choroba Niemanna-Picka). Głównym wskazaniem do diagnostyki biopsyjnej mięśnia sercowego jest choroba Fabry’ego. Jest to schorzenie związane z mutacją genu dla lizosomalnego enzymu α-galaktozydazy A, zlokalizowanego w chromosomie X. Skutkiem mutacji jest gromadzenie się glikosfingolipidów w lizosomach kardiomiocytów i ich stopniowa degeneracja. Degeneracja ta charakteryzuje się występowaniem nasilonej wakuolizacji komórek mięśniowych, jednak zmiany te są niespecyficzne i występują także w przewlekłych zmianach niedokrwiennych. Niezwykle pomocne okazuje się tutaj badanie wycinków w mikroskopie elektronowym, pozwalające na wykazanie obecności figur mielinowych w kardiomiocytach i komórkach śródbłonka naczyniowego.
Klinicznie choroba Fabry’ego przypomina kardiomiopatię przerostową27. Uważa się, że u około 3% mężczyzn z niejasnym etiologicznie przerostem mięśniówki lewej komory można rozpoznać homozygotyczną postać choroby Fabry’ego28. Od 2001 roku dostępna jest terapia substytucyjna rekombinowaną α-galaktozydazą (aglazydaza β). Po włączeniu leczenia kontrolna biopsja mięśnia sercowego jest wykorzystywana jako cenne narzędzie oceny odpowiedzi na lek29.
Diagnostyka biopsyjna w innych kardiomiopatiach wtórnych
Wśród wielu kardiomiopatii wtórnych typowanych do diagnostyki biopsyjnej na szczególną uwagę zasługują dwie z nich:
- kardiomiopatia antracyklinowa
- hemochromatoza.
Kardiomiopatia antracyklinowa jest następstwem zastosowania wielu leków z grupy antracyklinowej (doksorubicyna, daunorubicyna). Uszkodzenie miokardium zależy od wielkości dawki, a także od indywidualnej wrażliwości na te leki. Klinicznie kardiomiopatie antracyklinowe mogą przyjmować postać zarówno kardiomiopatii rozstrzeniowej, jak i restrykcyjnej. Uszkodzenie serca może mieć postać ostrej lub przewlekłej NS, pojawiającej się nawet po kilku latach od zaprzestania przyjmowania leków. Biopsja mięśnia sercowego jest traktowana jako najczulszy i specyficzny test uszkodzenia poantracyklinowego serca30. W ocenie histologicznej w wycinkach dominuje włóknienie zarówno śródmiąższowe, jak i zastępcze bez współistniejących cech przerostu kardiomiocytów. Jednak aby wykazać najwcześniejsze zmiany patologiczne, należy wykonać badanie elektronomikroskopowe wycinków tkankowych serca. Zgodnie z aktualnymi wskazaniami do biopsji endomiokardialnej diagnostyka biopsyjna powinna być rozważana w przypadku pojawienia się kardiomiopatii u leczonego chorego w kontekście możliwości utrzymania lub zwiększenia dawki leku15.
Hemochromatoza może być schorzeniem zarówno nabytym (liczne transfuzje krwi), jak i wrodzonym (mutacja genowa), występującym w następstwie gromadzenie się jonów żelaza w kardiomiocytach. Skutkiem tego jest rozwój niewydolności lewokomorowej serca. Do wykazania złogów żelaza w wycinkach endomiokardialnych wykorzystuje się barwienie histochemiczne błękitem pruskim31.
W pozostałych przypadkach kardiomiopatii pierwotnych i wtórnych należy kierować się indywidualnymi wskazaniami do takiej diagnostyki.