III Kongres Akademii po Dyplomie Okulistyka już w ten piątek! Kup bilet i dołącz do ekspertów podczas Siatkówka Meeting! Sprawdź >
NowoŚci w praktyce
Sprawozdanie z kongresu Amerykańskiego Towarzystwa Nefrologicznego Filadelfia 6-9 listopada 2008 r.
doc. dr hab. med. Beata Naumnik
prof. dr hab. med. Michał Myśliwiec

doc. dr hab. med. Beata Naumnik

prof. dr hab. med. Michał Myśliwiec
American Society of Nephrology (ASN) co roku organizuje spotkania służące podsumowaniu postępów w nefrologii i dziedzinach pokrewnych. W sprawozdaniu z ostatniego kongresu przekazujemy informacje istotne dla lekarzy praktyków i opisujemy nowości mogące znaleźć zastosowanie w praktyce.
Osteodystrofia nerkowa: nowe nazewnictwo i postępy w leczeniu
Podczas ostatniego kongresu ASN organizacja Kidney Disease: Improving Global Outcomes (KDIGO) zaproponowała zamianę nazwy „osteodystrofia nerkowa” na „mineralne i kostne powikłania przewlekłej choroby nerek” (chronic kidney disease related mineral and bone disorders). Obejmują one zmiany laboratoryjne, kostne i kalcyfikację tkanek miękkich, głównie naczyń. Zmiany laboratoryjne to odchylenia od normy w gospodarce wapniowo-fosforanowej, w aktywności fosfatazy alkalicznej i jej frakcji kostnej oraz zmiany innych parametrów metabolizmu kości. Zmiany kostne dotyczą metabolizmu, mineralizacji i objętości beleczek. Metabolizm i objętość określa się jako zmniejszone, prawidłowe lub zwiększone, a mineralizację jako prawidłową lub nieprawidłową. Trzy główne składowe mineralnych i kostnych powikłań przewlekłej choroby nerek występują samodzielnie bądź w różnych konfiguracjach, obejmując u części chorych zarówno zmiany laboratoryjne, kostne, jak i kalcyfikację tkanek miękkich.
Już w drugim stadium przewlekłej choroby nerek widoczna staje się tendencja do retencji fosforanów, ale zwiększenie stężenia czynnika wzrostu fibroblastów (FGF-23) powoduje zahamowanie wchłaniania zwrotnego fosforanów w cewkach nerkowych, dzięki czemu stężenie fosforu we krwi utrzymuje się w normie. Wczesnym wskaźnikiem zaburzonej równowagi fosforanowej w przewlekłej chorobie nerek jest zatem zwiększona utrata fosforanów z moczem i podwyższone stężenie FGF-23 we krwi. Niekorzystnym działaniem FGF-23 jest hamowanie hydroksylacji witaminy D w pozycji 1 i w efekcie zmniejszenie wytwarzania jej aktywnej formy – kalcytriolu. Zmniejszenie stężenia witaminy D i wapnia oraz wzrost stężenia fosforu we krwi nasila wytwarzanie parathormonu w przytarczycach i powoduje ich przerost (wtórna nadczynność przytarczyc). Parathormon nasila metabolizm kości i wywołuje ich włókniste zapalenie (osteitis fibrosa). Leczenie wtórnej nadczynności przytarczyc polega na ograniczeniu podaży fosforanów w diecie, hamowaniu ich wchłaniania w przewodzie pokarmowym (najczęściej przez podawanie węglanu wapnia), uzupełnianiu witaminy D, która hamuje wytwarzanie parathormonu, i podawaniu kalcimimetyków, które obniżają stężenie wapnia konieczne do hamowania wydzielania parathormonu. W przypadkach wtórnej nadczynności przytarczyc opornej na leczenie należy wykonać paratyreoidektomię.
Zmniejszony metabolizm kości, którego następstwem jest adynamiczna choroba kości (adynamic bone disease), stwierdza się u około połowy dializowanych chorych. Chorobę charakteryzuje obniżony metabolizm kości (resorpcja i tworzenie kości) ze zmniejszeniem liczby osteoklastów i osteoblastów oraz prawie całkowity zanik wytwarzania osteoidu, co można ocenić w biopsji po wcześniejszym znakowaniu tetracykliną. Adynamiczną chorobę kości należy różnicować z osteomalacją, polegającą na tworzeniu osteoidu, który nie ulega mineralizacji. Do niedawna przyczyną osteomalacji było zatrucie glinem wykorzystywanym do wiązania fosforanów w przewodzie pokarmowym. Obecnie unika się stosowania związków glinu, więc od ok. 15 lat częstość osteopatii glinowej stopniowo się zmniejsza. Za adynamiczną chorobą kości przemawia zmniejszone stężenie parathormonu we krwi, czemu towarzyszy obniżenie aktywności fosfatazy alkalicznej, zwłaszcza jej frakcji kostnej. U części chorych nawet znacznie podwyższone stężenie parathormonu nie wyklucza adynamicznej choroby kości. W przypadku obniżonej aktywności fosfatazy alkalicznej nie powinno się stosować kalcimimetyków, preparatów wapnia i witaminy D ani wykonywać paratyreoidektomii. Wysokie stężenie parathormonu i aktywności fosfatazy alkalicznej zdecydowanie przemawia za zwiększonym metabolizmem kości, będącym skutkiem nadczynności przytarczyc. Zmniejszenie bólów kostnych i tendencji do złamań po zastosowaniu preparatów hamujących wytwarzanie parathormonu potwierdza rozpoznanie. Kalcyfikacja tkanek miękkich, zwłaszcza naczyń (głównie kompleksu intima-media dużych tętnic i zastawek serca), może towarzyszyć zarówno adynamicznej chorobie kości, jak i włóknistemu zapaleniu kości we wtórnej nadczynności przytarczyc. W adynamicznej chorobie kości wapń i fosfor nie są zużywane do wytwarzania kości, zwiększa się więc ich stężenie we krwi, co sprzyja odkładaniu w tkankach miękkich fosforanu wapnia (a w konsekwencji hydroksyapatytu). Wykazano wyraźną korelację między zahamowaniem tworzenia kości a przemianą komórek mięśni gładkich tętnic w osteoblasty. Stopień kalcyfikacji naczyń jest odwrotnie proporcjonalny do metabolizmu kości i wprost proporcjonalny do obciążenia wapniem. Tworzeniu zwapnień w tętnicach sprzyjają: podeszły wiek, wieloletnia dializoterapia, niedożywienie, nasilenie nieswoistego zapalenia (objawiającego się m.in. wzrostem stężenia fibrynogenu we krwi), nadmierna podaż preparatów wapnia i witaminy D, adynamiczna choroba kości. Zwapnienia dużych tętnic powodują zwiększenie ich sztywności, czego skutkiem jest wzrost szybkości rozchodzenia się fali tętna, a to z kolei obciąża serce i sprzyja przerostowi lewej komory.
Adynamiczna choroba kości jest najczęściej chorobą jatrogenną, spowodowaną nadmierną podażą preparatów wapnia i witaminy D oraz kalcimimetyków stosowanych w celu hamowania wytwarzania parathormonu. W diagnostyce rodzaju zaburzeń mineralnych i kostnych należy nie tylko opierać się na aktualnych wynikach badań, ale także brać pod uwagę kierunek zmian w ciągu kilku lub kilkunastu tygodni, ponieważ ani nadmierne hamowanie wytwarzania parathormonu, ani tolerowanie jego nadprodukcji nie jest korzystne. W przypadku niejasnego obrazu klinicznego i trudności w interpretacji badań laboratoryjnych należy wykonać biopsję kości. U ok. 1/4 chorych nie da się jednak ustalić właściwego rozpoznania na tej podstawie, gdyż zarówno w adynamicznej chorobie kości, jak i nadczynności przytarczyc wyniki badania histopatologicznego kości są podobne. W takich przypadkach po kilku miesiącach ponownie należy wykonać biopsję i wówczas ocenić kierunek zmian po zastosowanym leczeniu.
Czynnikami ryzyka adynamicznej choroby kości są: podeszły wiek, cukrzyca, niedożywienie, zwiększona podaż preparatów wapnia i witaminy D, nadmierne hamowanie wydzielania parathormonu preparatami witaminy D i wapniowymi wiązaczami fosforanów w przewodzie pokarmowym. Może się ona rozwinąć u leczonych kalcitriolem mimo zwiększonego stężenia parathormonu we krwi. Przedstawione na kongresie prace nie potwierdziły, jakoby ta choroba rozwijała się częściej u dializowanych otrzewnowo. Wydaje się, że zależy to głównie od rodzaju podawanych leków.
Chorym z adynamiczną chorobą kości nie należy podawać preparatów wapnia i witaminy D ze względu na wywoływanie przez nie hiperkalcemii i możliwość nadmiernego hamowania wydzielania parathormonu. Z uwagi na niebezpieczeństwo nadmiernego hamowania wydzielania parathormonu nie należy też stosować kalcimimetyków, zwłaszcza u chorych z obniżoną aktywnością fosfatazy alkalicznej. Do wiązania fosforanów w przewodzie pokarmowym powinno się wykorzystywać preparaty niezawierające wapnia, takie jak sewelamer i węglan lantanu. Ich stosowanie nie wiąże się ze stanami hiperkalcemii, które predysponują do kalcyfikacji naczyń. Sewelamer i węglan lantanu wiążą fosforany w stopniu podobnym do węglanu wapnia, obniżając dodatkowo stężenie cholesterolu całkowitego i LDL o 30-35% i jednocześnie zwiększając stężenie cholesterolu HDL o 10-30%.
W stanach zwiększonego lub normalnego metabolizmu kości kalcyfikacja naczyń tylko w niewielkim stopniu zależy od podaży wapnia. W adynamicznej chorobie kości kalcyfikacja aorty jest wprost proporcjonalna do obciążenia wapniem. W kalcyfikacji odgrywają także rolę jej promotory (osteokalcyna, wysokie stężenie fosforu i wapnia, białko morfogenetyczne kości BMP-2, osteonektyna, leptyna, fosfataza alkaliczna, kolagen typu I, fibronektyna, oksydowany LDL, deksametazon, PTH 7-84, czynnik martwicy nowotworów, niedobór białka Klotho, onkostatyna,) i inhibitory (fetuina-A, białko morfogenetyczne kości BMP-7, osteoprotegeryna, pirofosforany, białko macierzy [MGP – matrix-Gla protein], osteopontyna, kolagen typu IV, PTH 1-34, receptor PTH).
Postępowanie w adynamicznej chorobie kości
- Unikać nadmiaru preparatów wapnia i witaminy D, zwłaszcza jej form aktywnych
- Unikać nadmiaru wapnia w płynie dializacyjnym (obniżyć stężenie wapnia do 1,25 mmol/l)
- Kontrolować stężenie PTH i wapnia we krwi
- Nie hamować wydzielania PTH ani nie wykonywać paratyreoidektomii
- U niedożywionych zastosować leczenie żywieniowe
- W przypadku niejasności wykonać biopsję kości
Ostra niewydolność nerek: nowe nazewnictwo i wczesne wskaźniki ostrego uszkodzenia nerek
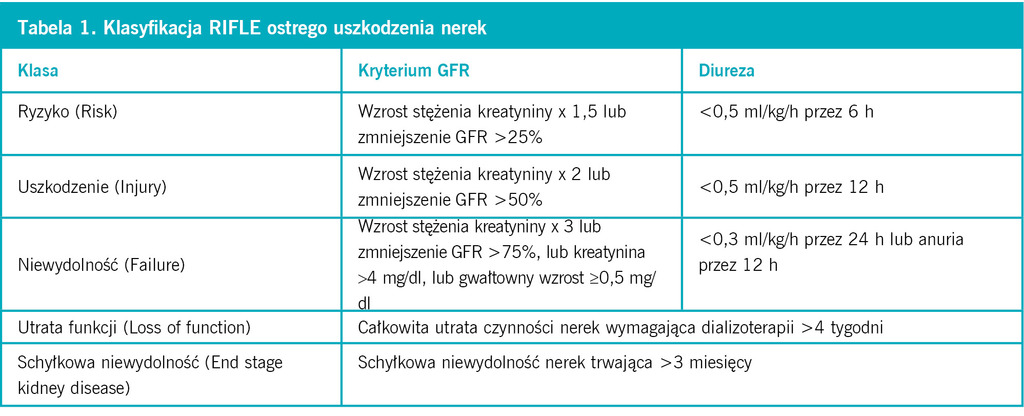
Tabela 1. Klasyfikacja RIFLE ostrego uszkodzenia nerek
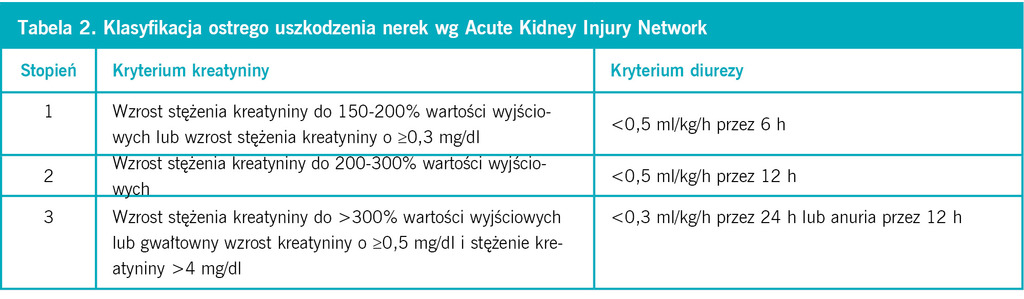
Tabela 2. Klasyfikacja ostrego uszkodzenia nerek wg Acute Kidney Injury Network
Ostra niewydolność nerek jest końcowym stadium ostrego uszkodzenia nerek, a do wzrostu stężenia kreatyniny dochodzi bardzo późno, gdy zniszczone jest około połowy miąższu nerki. Wiadomo, że im wcześniej rozpozna się ostre uszkodzenie nerek, tym skuteczniejsze jest leczenie i zapobieganie postępowi ich niewydolności. Definicja ostrego uszkodzenia nerek wciąż opiera się na zwiększeniu stężenia kreatyniny we krwi (o ponad 0,3 mg/dl lub o ≥50%) lub spadku diurezy (<0,5 ml/kg/h przez ≥6 h). Dotąd, zgodnie z klasyfikacją RIFLE (Risk, Injury, Failure, Loss of function and End stage kidney disease), ostre uszkodzenie nerek dzielono na 5 klas – od zadziałania czynnika ryzyka do schyłkowej niewydolności. Teraz wprowadzono podział na 3 stopnie w zależności od stopnia wzrostu stężenia kreatyniny lub zmniejszenia diurezy. Stopnie praktycznie odpowiadają pierwszym trzem klasom RIFLE, pominięto natomiast dwie ostatnie uwzględniające rokowanie (tab. 1 i 2). Poszukuje się ciągle możliwości szybszej diagnostyki ostrego uszkodzenia nerek, zanim rozwinie się niewydolność, którą trzeba leczyć dializami. Wczesnymi wskaźnikami ostrego uszkodzenie nerek jest wzrost stężenia w moczu białka NGAL (neutrophil gelatinase-associated lipocalin – białko związane z żelatynazą neutrofilów) o masie 25 kDa. Podwyższa się ono w ciągu 2 h po zadziałaniu czynnika toksycznego, ma blisko 100% specyficzność i czułość, dlatego nazywane jest troponiną uszkodzenia nerek. Inne białko, którego stężenie w moczu jest zwiększone wcześnie po zadziałaniu bodźca (2-4 h), to KIM-1 (kidney injury molecule-1). Wcześnie wzrasta też stężenie w moczu interleukiny 18, które również pozostaje podwyższone przez kilkadziesiąt godzin. Poznano dotąd ok. 30 białek, będących wczesnymi wskaźnikami ostrego uszkodzenia nerek. Pochodzą one z osocza, komórek cewek lub naciekających komórek zapalnych. Ich znaczenie kliniczne wymaga jednak szerszych badań, z użyciem niezamrożonych próbek moczu i krwi.
Zapalenie drobnych naczyń (vasculitis) oporne na leczenie
Podstawowym schematem indukcji remisji w zapaleniu drobnych naczyń (vasculitis) pozostają glikokortykosteroidy z cyklofosfamidem. Przy gwałtownym postępie choroby można wykonać plazmaferezę. W indukcji remisji nigdy nie powinno się stosować glikokortykosteroidów w monoterapii. Zalecane dawkowanie cyklofosfamidu to pulsy (15 mg/kg/puls co 3 tygodnie) lub terapia doustna 2 mg/kg/24h. Leczenie cyklofosfamidem nie powinno trwać krócej niż 6 miesięcy, ale przy braku cech niewydolności nerek i po uzyskaniu szybkiej remisji terapię można zakończyć po 3 miesiącach. Jeżeli wystąpi oporność na schemat podstawowy (po 6 miesiącach brak remisji), terapią drugiego wyboru jest dodanie do glikokortykosteroidów mykofenolanu mofetilu lub leflunomidu. W przypadku niepowodzenia można podać cyklosporynę A z azatiopryną. Jeśli mimo zastosowania wymienionych schematów nie następuje poprawa, można podawać metotreksat w dawce 25 mg na tydzień. Ostatnim wyborem jest rituksimab w połączeniu z pulsami cyklofosfamidu lub mykofenolanu mofetilu (3 × 1 g). W leczeniu stabilizującym remisję wykorzystuje się glikokortykosteroidy w dawkach podtrzymujących, które można stosować do 18 miesięcy. Nie należy jednak odstawiać ich zbyt szybko. Aby podtrzymać remisję, można także podawać azatioprynę (nawet w monoterapii). Taką terapię można prowadzić nawet do 4 lat.
Prawdopodobieństwo nawrotu zapalenia drobnych naczyń jest większe u kobiet i osób starszych oraz w przypadku zajęcia płuc, stosowania metotreksatu w indukcji i dużej dawki cyklofosfamidu potrzebnej do uzyskania remisji. Samo wykrycie przeciwciał przeciw cytoplazmie neutrofilów (ANCA – anti-neutrophil cytoplasmic antibodies) w surowicy nie jest wskazaniem do podjęcia terapii.