III Kongres Akademii po Dyplomie Okulistyka już w ten piątek! Kup bilet i dołącz do ekspertów podczas Siatkówka Meeting! Sprawdź >
Pulmonologia
Mukowiscydoza – choroba (nie)rzadka
dr n. med. Hanna M. Winiarska
lek. Filip Wojtaś

dr n. med. Hanna M. Winiarska

lek. Filip Wojtaś
- Postępy w leczeniu mukowiscydozy – wprowadzenie modulatorów CFTR
- Wpływ choroby na pracę różnych układów i narządów, a także na ogólny dobrostan pacjentów
- Znaczenie kompleksowej opieki i edukacji pacjentów oraz wyzwania wynikające z wydłużenia życia chorych na mukowiscydozę
Mukowiscydoza (CF – cystic fibrosis), zwana najczęstszą chorobą rzadką, jest chorobą genetyczną, dziedziczoną autosomalnie recesywnie, występującą głównie u rasy kaukaskiej. Istotą patologii jest mutacja przezbłonowego kanału chlorkowego (CFTR – cystic fibrosis transmembrane conductance regulator), która prowadzi do zagęszczenia wydzielanego śluzu, a w konsekwencji do uszkodzenia wielu narządów, przede wszystkim układu oddechowego (oskrzeli, zatok obocznych nosa), ale także trzustki (początkowo tylko zewnątrz-, ale po latach także jej wewnątrzwydzielniczej funkcji). Ponadto dochodzi do powstania niedrożności smółkowej (objaw patognomoniczny), zaburzeń wchłaniania tłuszczów i witamin w nich rozpuszczalnych oraz układu rozrodczego (nasieniowody, jajowody).
W Polsce liczba pacjentów z mukowiscydozą wynosi około 2000, z czego blisko 1/3 to osoby dorosłe. Według danych Głównego Urzędu Statystycznego z 2024 roku w Polsce jest około 10 000 specjalistów medycyny rodzinnej. To oznacza, że co 5 specjalista medycyny rodzinnej będzie miał wśród swoich stałych pacjentów chorego z mukowiscydozą, a spodziewane wydłużenie życia oraz poprawa w zakresie zdrowia prokreacyjnego mogą spowodować zwiększenie populacji chorych wymagających szczególnej opieki. Niniejszy artykuł jest próbą usystematyzowania wiedzy na temat tej choroby, opisania nowych możliwości terapeutycznych i ich konsekwencji dla życia pacjentów, ale także dla zmieniającego się obrazu schorzenia.
W Polsce od lipca 2009 roku u noworodków są wykonywane badania przesiewowe w kierunku mukowiscydozy. W 3 dobie życia pobiera się krew z pięty dziecka na bibułę w celu oznaczenia stężenia immunoreaktywnej trypsyny. W przypadku uzyskania dodatniego wyniku dzieci są kierowane na dalszą diagnostykę, która obejmuje badanie genetyczne pod kątem najczęstszych w populacji polskiej mutacji białka CFTR. U dzieci z mutacjami na obu allelach rozpoznaje się mukowiscydozę i obejmuje je specjalistyczną opieką. Dzieci z mutacją 1 allelu są kierowane także do specjalistycznego ośrodka, w którym między innymi oznacza się chlorki w pocie. Wynik tego badania determinuje ewentualne rozpoznanie mukowiscydozy przy niekonkluzywnym wyniku badań genetycznych.
Z uwagi na wysoką czułość badań przesiewowych powstaje populacja dzieci, u których nie rozpoznano mukowiscydozy mimo nieprawidłowych wyników badania przesiewowego, po przeprowadzeniu pogłębionych badań genetycznych (obejmujących większą liczbę mutacji niż badania klasyczne) lub uzyskaniu niekonkluzywnego wyniku testu potowego. Takich pacjentów określa się mianem CFSPID (Cystic Fibrosis Screen Positive, Inconclusive Diagnosis). To pacjenci, którzy powinni pozostać pod obserwacją ośrodka pediatrycznego i być monitorowani pod kątem ujawnienia się typowych dla mukowiscydozy objawów.
Badania przesiewowe obejmujące niemal całą populację noworodków w Polsce sprawiły, że choroba jest rozpoznawana na bardzo wczesnym, zwykle bezobjawowym etapie. Takie dziecko zaś od początku jest objęte opieką specjalistyczną, odpowiednią suplementacją odżywczą, ewentualną fizjoterapią bądź leczeniem mukolitycznym. Postępowanie to opóźnia nieuchronną destrukcję wielonarządową, poprawia jakość i długość życia chorych oraz umożliwia szybkie wykrywanie i leczenie powikłań.
Kolejnym krokiem milowym (żeby nie powiedzieć „rewolucyjnym”) w leczeniu mukowiscydozy było wprowadzenie w 2022 roku w ramach programu Narodowego Funduszu Zdrowia (NFZ) tzw. modulatorów białka CFTR, ogólnie nazywanych także lekami przyczynowymi. Terapia w Polsce została wdrożona zaledwie 3 lata po pierwszej rejestracji leków w USA. Nasi chorzy mają zatem dostęp do najnowocześniejszego leczenia na światowym poziomie.
Aby zrozumieć, czym są modulatory białka CFTR, należy krótko zagłębić się w genetykę samej choroby. Mutacja białka CFTR może oznaczać powstanie białka o charakterze nonsensownym (zupełnie niedziałającego, wówczas modulatory nie są skuteczne) albo takiego, które wykazuje zmniejszoną funkcję, upośledzony transport na powierzchnię błony komórkowej lub przyspieszony rozpad. Na te właśnie białka wpływają modulatory, które poprawiają ich funkcjonowanie. Spośród leków dostępne są tak zwane potencjalizatory (iwakaftor) oraz korektory (lumakaftor, tezakaftor, eleksakaftor). Dobór leków stosowanych u konkretnego pacjenta zależy od jego wieku oraz mutacji białka CFTR. Nie jest to leczenie przyczynowe w dosłownym tego słowa znaczeniu (gen nadal pozostaje uszkodzony), a niejako substytucyjne, umożliwiające lepsze funkcjonowanie nieprawidłowego białka. Wprowadzenie leków wywołało rewolucję w stanie chorych z mukowiscydozą. Pacjenci początkowo zaczęli odkrztuszać zwiększoną ilość wydzieliny, ale już po kilku dniach lub tygodniach jej objętość znacznie się zmniejszyła (niektórzy nie wykrztuszają już wcale). W związku z tym chorzy odczuwają mniejszą duszność, mniej kaszlą, mają istotnie mniej zaostrzeń. Można u nich także zaobserwować poprawę w zakresie parametrów spirometrycznych, szczególnie nasilonej pierwszosekundowej objętości wydechowej (FEV1 – forced expiratory volume in one second), której spadek jest jednym z najistotniejszych czynników predykcyjnych zgonu u tych chorych. U pacjentów widoczny jest także wzrost wskaźnika masy ciała (BMI – body mass index), nierzadko prowadzący nawet do otyłości. U chorych, u których nie doszło jeszcze do całkowitego uszkodzenia miąższu trzustki, można odnotować znamienną poprawę jej funkcji.
W związku z istotną zmianą obrazu choroby konieczne jest ponowne wyedukowanie specjalistów medycyny rodzinnej tak, aby nadal stanowili silnych partnerów w leczeniu pacjentów z mukowiscydozą. Poniżej zostaną przedstawione główne aspekty, na które należy zwrócić uwagę u chorych poddanych terapii modulatorami białka CFTR.
Układ oddechowy
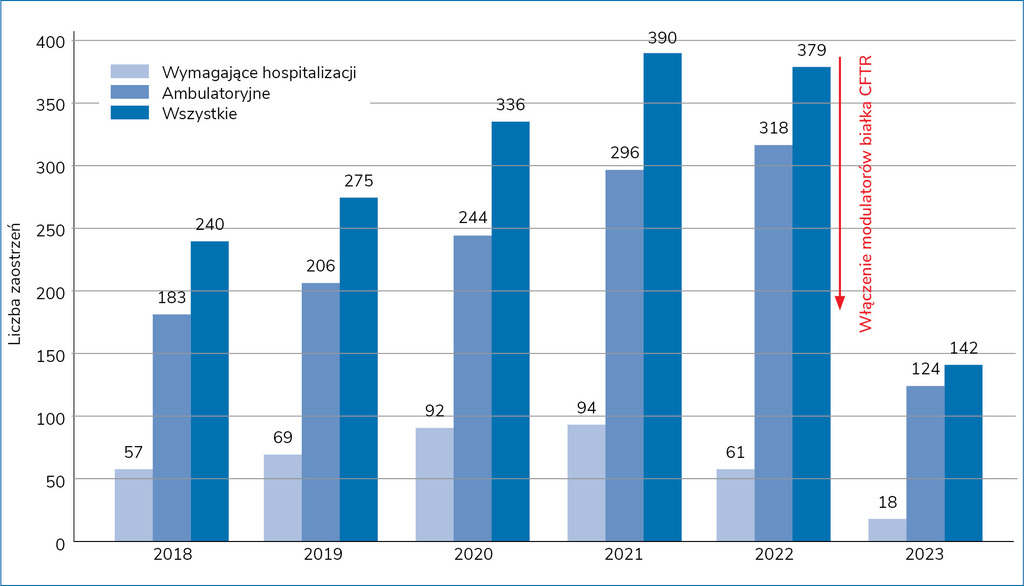
Rycina 1. Liczba zaostrzeń mukowiscydozy w ciągu roku na Oddziale Pulmonologii Uniwersyteckiego Szpitala Klinicznego w Poznaniu

Tabela 1. Kryteria Fuchsa dotyczące rozpoznania zaostrzenia mukowiscydozy2
Dzięki wprowadzeniu modulatorów białka CFTR zmniejszyła się częstość zaostrzeń choroby podstawowej, co (na przykładzie poznańskiego ośrodka) przedstawiono na rycinie 11. Pacjenci odkrztuszają mniej lub wcale, drenaż wydzieliny z oskrzeli bywa zerowy. Dlatego zasadne jest odstawianie u tych pacjentów leków mukolitycznych (szczególnie tych o charakterze drażniącym – hipertoniczna sól). U chorych, którzy w ogóle nie odkrztuszają, po analizie zysków i strat można także ostrożnie odstawić dornazę α bądź pozostałe leki o podobnej funkcji. Warto jednak pamiętać o konieczności niezwłocznego ponownego włączenia mukolityków w przypadku wystąpienia zaostrzenia. W związku z tym, że zaostrzenia bywają lżejsze, chorzy w pierwszej kolejności trafią do lekarza rodzinnego. Nadal obowiązuje kilkudziesięcioletnia już definicja zaostrzenia w mukowiscydozie, mówiąca o pojawieniu się co najmniej 4 z 12 kryteriów zaostrzenia według Fuchsa (szczegóły podano w tabeli 1)2.

Tabela 2. Antybiotykoterapia empiryczna w zaostrzeniu mukowiscydozy3
Każde zaostrzenie powinno być leczone antybiotykami, zgodnie z najnowszym posiewem plwociny/wymazu z nosa i gardła/bronchofiberoskopii. W przypadku gdy wyniki ostatnich posiewów (szczególnie u pacjentów z nieproduktywnym kaszlem, mających tylko wyniki badania mikrobiologicznego z górnych dróg oddechowych) są ujemne, warto sięgnąć do ostatniego dodatniego posiewu albo zastosować antybiotykoterapię empiryczną przedstawioną w tabeli 23. Pacjenci z mukowiscydozą są przewlekle skolonizowani bakteryjnie, a antybiotykoterapia nie jest w stanie doprowadzić do eradykacji drobnoustrojów. W związku z tym zakłada się, że zaostrzenie choroby zwykle wynika z namnożenia się bakterii przewlekle rezydujących w dolnych drogach oddechowych, i dlatego z powodzeniem można się odnosić do antybiogramu nawet sprzed kilku miesięcy.
Warto sięgnąć po dawkowanie antybiotyków zamieszczone w wytycznych KOMPAS CF z 2017 roku4. Czas trwania antybiotykoterapii w zaostrzeniu mukowiscydozy to co najmniej 10, a w niektórych przypadkach nawet 14 dni. Dawki leków stosowanych w zaostrzeniu są wyższe niż w populacji ogólnej. Należy jednak zwrócić uwagę na interakcję niektórych leków z modulatorami białka CFTR. Dotyczy to głównie tych leków, których metabolizm odbywa się za pomocą cytochromu CYP3A4. Wówczas konieczne jest zmodyfikowanie dawkowania modulatorów zgodnie z tabelą zamieszczoną w charakterystyce produktu leczniczego.
Jeżeli nie jest możliwe przeleczenie pacjenta lekami doustnymi/wziewnymi, wymagany jest kontakt z ośrodkiem referencyjnym, który zadecyduje o ewentualnym przyjęciu chorego do szpitala na antybiotykoterapię dożylną lub o wdrożeniu finansowanego przez NFZ programu antybiotykoterapii domowej, w którym leki podaje przeszkolona pielęgniarka w domu pacjenta. Leki w ramach opisywanego programu są wydawane dla pacjenta ze szpitala za darmo, przewidziana jest także opłata za usługę pielęgniarską.