



Spis treści
STRESZCZENIE
Dystonie to heterogenna grupa chorób układu pozapiramidowego o zróżnicowanym obrazie klinicznym i różnej etiologii, charakteryzujących się występowaniem niekontrolowanych kurczów mięśni prowadzących do wymuszonego ułożenia części ciała. Istotną grupę wśród dystonii stanowią postacie genetycznie uwarunkowane. Do tej pory z występowaniem dystonii powiązano 25 loci chromosomowych i zidentyfikowano 16 genów, których mutacje są odpowiedzialne za jej różne postacie. Zidentyfikowane
geny kodują m.in. białka związane ze szlakiem syntezy dopaminy, przewodnictwem synaptycznym, funkcjonowaniem cytoszkieletu, detoksykacją czy metabolizmem energetycznym komórki, co tłumaczyć może tak zróżnicowany patomechanizm i obraz kliniczny dystonii.
Wprowadzenie
Dystonie to termin określający nieprawidłowe, niezależne od woli, powtarzające się niekontrolowane ruchy, występujące w różnych częściach ciała, prowadzące do wymuszonego ich ułożenia. Termin ten oznacza także bardzo heterogenną grupę chorób neurologicznych związanych z zaburzeniami funkcjonowania jąder podstawy, co skutkuje dominacją ruchów dystonicznych w ich obrazie klinicznym.
Dystonie najczęściej występują sporadycznie, opisano jednak wiele rzadkich postaci rodzinnych o mendlowskim sposobie dziedziczenia, a dotąd zidentyfikowano 16 genów zaangażowanych w molekularne podłoże tej choroby. 1 Pojęcie dystonii wprowadził Herman Oppenheim, który w 1911 r. opisał dystonię mięśniową deformacyjną (obecnie dystonia torsyjna typu 1), chociaż kluczowe opisy dla zrozumienia systematyki oraz rzucające światło na patogenezę przyniosły badania Sterlinga i Flauta. 2, 3 Podłoże molekularne, a zarazem pierwszy gen, którego mutacje powiązano z dystonią, zidentyfikowano jednak dopiero w roku 1994. 4 Genetycznie uwarunkowane dystonie są chorobami rzadkimi, co w znacznym stopniu utrudnia badania. Brakuje dostatecznie licznych grup pacjentów jednorodnych pod względem klinicznym, co sprawia, że wykorzystanie i skuteczność klasycznych metod badawczych, takich jak analiza sprzężeń czy badania asocjacji, są ograniczone. W ostatnich latach rozwój technik sekwencjonowania DNA przyczynił się do znacznego postępu w poznaniu patologii molekularnej dystonii.
Celem prezentowanej pracy jest omówienie i charakterystyka podłoża molekularnego oraz patologii różnych postaci genetycznie uwarunkowanych dystonii.
Klasyfikacja dystonii
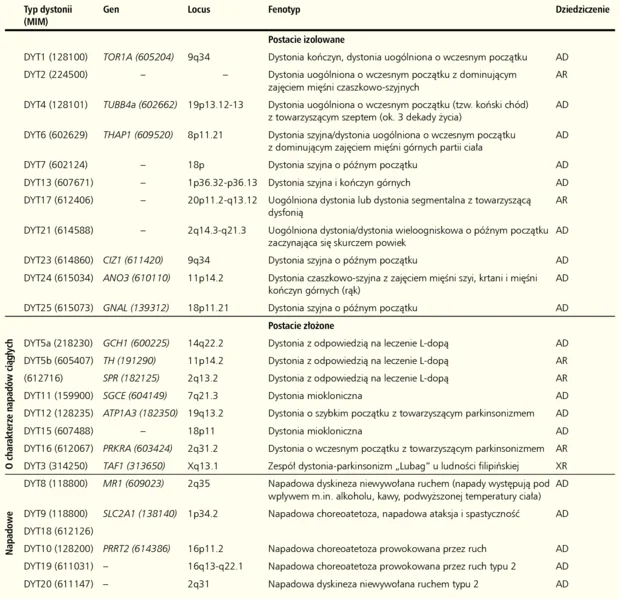
Tabela. Klasyfikacja molekularna dystonii uwarunkowanych genetycznie
Postacie izolowane dystonii
Sporadyczne postacie izolowane dystonii stanowią grupę różnych chorób, w których dystonia jest jedynym objawem, a podłoże nie jest wynikiem procesu neurodegeneracyjnego. Do tej rodziny zaliczamy sześć genetycznie uwarunkowanych dystonii – dystonia torsyjna typ 1 (DYT1) oraz dystonie typu 4, 6, 23, 24 i 25.
Dystonia torsyjna typu 1
Dystonia torsyjna typu 1 (DYT1) jest dominującą postacią dystonii okresu rozwojowego. Pierwsze jej objawy mogą pojawić się w okresie dojrzewania, wieku młodzieńczym, a w sporadycznych przypadkach u dorosłych, zarówno pod postacią dystonii uogólnionej, jak i dystonii ogniskowej/segmentalnej, początkowo dotyczącej kończyny górnej albo dolnej, z czasem przyjmujące postać uogólnioną. 7 Obraz kliniczny choroby może zmieniać się w czasie w zależności od wieku pacjenta, manifestacji objawów i progresji choroby. Choroba dziedziczy się w sposób autosomalny dominujący (AD) z penetracją wynoszącą 30-40%, 8 a jej podłoże molekularne stanowią mutacje genu TOR1A. Gen ten koduje białko torsynę A (TOR1A), zlokalizowane w zakończeniach dendrytycznych i aksonalnych neuronów, gdzie prawdopodobnie bierze udział w zachowaniu integralności cytoszkieletu, transporcie aksonalnym i recyklingu pęcherzyków synaptycznych. 9, 10, 11 Do tej pory opisano jedną patogenną mutację genu TOR1A w pozycji c.934-936GAGdel, skutkującą delecją jednego aminokwasu – kwasu glutaminowego (ΔE302/303) w białku i zaburzającą proces tworzenia funkcjonalnych oligomerów torsyny A. 12 Patogenny charakter kilku innych mutacji zidentyfikowanych w tym genie pozostaje nadal niewyjaśniony. 13, 14, 15, 16
Dystonia typu 4
Dystonia typu 4 (DYT4) charakteryzuje się późniejszym wiekiem zachorowania (około trzeciej dekady życia), a jej głównym objawem jest dystonia uogólniona z współwystępującymi zaburzeniami chodu (tzw. koński chód) oraz dysfonią. 17 Choroba dziedziczona jest w sposób autosomalny dominujący, a jej podłoże stanowią mutacje genu TUBB4 kodującego tubulinę 4β, podjednostkę składową mikrotubul. 18 Zmutowane białko może powodować destabilizację cytoszkieletu przez niekorzystny wpływ na proces tworzenia i strukturę mikrotubul. 19 TUBB4 ulega silnej ekspresji w mózgu, szczególnie zaś w móżdżku, którego dysfunkcja wiązana jest z występowaniem objawów dystonii. 18
Dystonia typu 6
Dystonia typu 6 (DYT6) może ujawnić się między 4 a 55 rokiem życia. Ruchy dystoniczne w tej postaci choroby dotyczą najczęściej mięśni unerwianych przez nerwy czaszkowe, mięśnie szyi, krtani oraz kończyn górnych i mają tendencję do rozszerzania się na inne części ciała. U pacjentów często występuje dysfonia. W większości przypadków choroba dziedziczy się w sposób autosomalny dominujący z penetracją na poziomie około 60%. 20 Podłoże molekularne stanowią w tym przypadku mutacje w genie THAP1. Białko kodowane przez ten gen wiąże się z DNA i bierze udział w regulacji transkrypcji własnego genu i wielu innych. Jednym z możliwych patomechanizmów DYT6 jest zaburzenie regulacji genów ulegających ekspresji w mózgu. 21
Dystonia typu 23
Dystonia typu 23 (DYT23) ma postać dystonii ogniskowej z zajęciem mięśni szyi, której towarzyszyć może lekkie drżenie. Choroba ujawnia się we wczesnym dzieciństwie i w wieku dojrzałym, dziedziczona jest autosomalnie dominująco. Podłoże molekularne choroby najprawdopodobniej stanowią mutacje w genie CIZ1. 22 Mutacje w tym genie zostały powiązane z występowaniem dystonii przez zastosowanie połączonych metod analizy sprzężeń i sekwencjonowania następnej generacji (next generation sequensing, NGS). 23 Dla jednoznacznego potwierdzenia udziału mutacji genu CIZ1 w patogenezie dystonii wymagane są jednak badania na większej grupy pacjentów. Gen CIZ1 koduje białko zaangażowane w syntezę DNA i kontrolę cyklu komórkowego. Silną jego ekspresję zaobserwowano w neuronach ruchowych i móżdżku. 22
Dystonia typu 24
Dystonia typu 24 (DYT24) ujawnia się we wczesnym dzieciństwie, ale także u dorosłych. Ma postać dystonii ogniskowej/segmentalnej obejmującej mięśnie twarzy, szyi, kończyn górnych i krtani z dominującym drżeniem głowy i rąk. Niekiedy jedynym jej objawem jest drżenie kończyn górnych mylnie klasyfikowane jako drżenie samoistne. Za kliniczną manifestację objawów odpowiedzialne są mutacje w genie ANO3, podobnie jak CIZ1 zidentyfikowanym za pomocą analizy sprzężeń i NGS. Gen ten koduje białko anoktaminę 3 o niezbyt dobrze poznanej funkcji, najprawdopodobniej zaangażowane w utrzymanie stężeniu jonów Ca2+ w komórce. 24 Biorąc pod uwagę fakt, że ulega on silnej ekspresji w prążkowiu, a kanały aktywowane jonami Ca2+ modulują pobudliwość neuronalną jest możliwe, że mutacje w tym genie prowadzą do nieprawidłowej pobudliwości neuronów prążkowia.
Dystonia typu 25
Dystonia typu 25 (DYT25) klinicznie przypomina DYT6. Ruchy dystoniczne obejmują przede wszystkim mięśnie szyi, twarzy, krtani, czasem mięśnie kończyn dolnych. Nie obserwuje się zajęcia mięśni kończyn górnych. Choroba ujawnia się w wieku dojrzałym, dziedziczona jest jako cecha autosomalnie dominująca. W 2012 roku zidentyfikowany został gen GNAL, którego mutacje odpowiedzialne są za tę postać dystonii. 25 Gen ten koduje białko Gαolf, którego nieprawidłowe funkcjonowanie może zakłócać przekaźnictwo dopaminergiczne. 26, 27
Dystonie złożone
Dystonie złożone scalają te postaci choroby, gdzie dystonii towarzyszą inne zaburzenia ruchowe, takie jak mioklonia lub parkinsonizm oraz okresowo występujące nieprawidłowości ruchowe, między którymi nie stwierdza się żadnych zaburzeń neurologicznych. Do tej grupy należą dystonie typu 3, 11, 12 i 16. 6
Dystonia typu 3
Dystonia typu 3 (DYT3) objawia się w wieku dojrzałym jako dystonia ogniskowa, rozszerzając się następnie na pozostałe części ciała. W niektórych przypadkach objawami jej towarzyszącymi może być parkinsonizm i/lub drżenie. 28 Choroba dziedziczona jest w sposób recesywny sprzężony z chromosomem X 29 i związana z występowaniem mutacji w genie TAF1, które mogą prowadzić do obniżenia specyficznej jego ekspresji i ekspresji receptora dopaminy D2 w jądrze ogoniastym. 30
Dystonia typu 11
Charakterystycznym objawem dystonii typu 11 (DYT11) są ruchy miokloniczne obejmujące głównie ramiona i mięśnie osiowe, którym często towarzyszy dystonia ogniskowa lub segmentalna poprzedzona takimi objawami, jak parestezje w zajętych częściach ciała. Początek choroby przypada na pierwszą lub drugą dekadę życia, a pacjenci często poza zaburzeniami motorycznymi cierpią na depresję, napady lęku panicznego, nadpobudliwość ruchową oraz zachowania obsesyjno-kompulsywne. 31 Nasilenie objawów może zmniejszać się po spożyciu alkoholu. 32 Choroba powodowana jest obecnością mutacji w genie SGCE 31 o wysokiej ekspresji w ośrodkowym układzie nerwowym. Gen SGCE koduje białko – epsilon sarkoglikan o prawdopodobnie synaptycznej lokalizacji. 33 Jest to białko przezbłonowe wchodzące wraz z innymi podjednostkami sarkoglikanu w skład kompleksu dystrofina-glikoproteina obecnego w mięśniach i mózgu. Mutacje mogą prowadzić do zmiany lokalizacji białka z błony komórkowej do cytoplazmy, przyczyniając się do jego przedwczesnej degradacji. 34 DYT11 dziedziczona jest autosomalnie dominująco, a w 10-15% przypadków z ograniczoną penetracją zmutowanego allelu, jeżeli ma on pochodzenie matczyne. 35 Jest to wynikiem preferencyjnej inaktywacji allelu matczynego przez metylację tzw. wysp CpG regionu promotorowego genu. 36
Dystonia typu 12
W przypadku dystonii typu 12 (DYT12) ruchy dystoniczne, którym towarzyszy parkinsonizm pojawiają się nagle w 2-3 dekadzie życia. Obejmują kończyny górne i twarz, powodując często dyzartrię i dysfagię. Objawy mogą nasilać się pod wpływem stresu, gorączki, podczas wykonywania ćwiczeń fizycznych. 37 Choroba dziedziczy się w sposób autosomalny dominujący z obniżoną penetracją na poziomie 90%. 38 Za wystąpienie objawów odpowiedzialne są mutacje w ulegającym ekspresji wyłącznie w tkance nerwowej genie ATP1A3 kodującym podjednostkę pompy sodowo-potasowej zależnej od ATP. Mechanizm powodujący, że mutacje w tym genie prowadzą do nagłego wystąpienia choroby nie został wyjaśniony. Mutacje genu ATP1A3 mogą zaburzyć gradient Na+/K+ istotny dla pobudliwości neuronalnej, transportu neurotransmiterów i innych funkcji warunkujących prawidłowe przekaźnictwo impulsów nerwowych. 39 Mutacje de novo w ATP1A3 mogą być również przyczyną naprzemiennej hemiplegii dziecięcej (alternating hemiplegia of childhood, AHC), charakteryzującej się nawracającymi napadami porażenia połowiczego, zaburzeniami ruchowymi (napady toniczne, dystoniczne, choreoatetyczne) i postępującym opóźnieniem rozwoju psychomotorycznego. 40
Dystonia typu 16
Objawy kliniczne dystonii typu 16 (DYT16) ujawniają się we wczesnym dzieciństwie i okresie dojrzewania. Ruchy dystoniczne obejmujące kończyny ewoluują w postać uogólnioną. Charakterystyczną cechą kliniczną jest współistniejący parkinsonizm. Choroba dziedziczona jest autosomalnie recesywnie (AR). 41 Podłożem molekularnym DYT16 są mutacje genu PRKRA kodującego uwalniany w warunkach stresu komórkowego aktywator kinazy PKR, zaangażowanej w transdukcję sygnału, różnicowanie i proliferację komórek oraz apoptozę. 42
Dystonia odpowiadająca na L-DOPĘ
U podłoża molekularnego dystonii z odpowiedzią na leczenie L-DOPĄ leżą patogenne zmiany w genach kodujących enzymy niezbędne do biosyntezy neuroprzekaźnika – dopaminy. Do tej grupy chorób należą dwie jednostki chorobowe DYT5 oraz dystonia spowodowaną niedoborem reduktazy sepiapteryny.
Dystonia typu 5 obejmuje dwa podtypy DYT5a i DYT5b, wyróżniane na podstawie sposobu dziedziczenia i lokalizacji genetycznej. Objawy kliniczne tej dystonii pojawiają się zwykle między 4 a 10 rokiem życia. Najczęściej zajęte są mięśnie kończyny dolnej, następnie tułów, kończyny górne, szyja. Ruchom dystonicznym często towarzyszą objawy zespołu parkinsonowskiego. Charakterystyczną cechą choroby jest dobowa fluktuacja objawów, nasilenie ruchów dystonicznych w ciągu dnia. 43 Kliniczna manifestacja objawów jest tutaj skutkiem niedostatecznej syntezy lewodopy, prekursora dopaminy. Jeżeli choroba dziedziczona jest w sposób autosomalny dominujący, 44 za wystąpienie jej objawów odpowiedzialne są mutacje genu GCH1 kodującego cyklohydrolazę 1 – enzym katalizujący m.in. pierwszy etap syntezy dopaminy. Jest to podtyp DYT5a. Przyczyną wystąpienia dystonii typu DYT5b są natomiast mutacje w genie TH. Objawy kliniczne są bardzo podobne do DYT5a, jednak choroba dziedziczona jest w sposób autosomalny recesywny. 45 Gen TH koduje hydroksylazę tyrozyny, enzym zaangażowany w konwersję tyrozyny do lewodopy odgrywający kluczową rolę w funkcjonowaniu neuronów dopaminergicznych. 46
Postacie dystonii z odpowiedzią na lewodopę obejmują również inne rzadkie rodzaje chorób związanych z zaburzeniem szlaku syntezy dopaminy, z których najczęstszą postacią jest dystonia z dobrą odpowiedzią na lewodopę spowodowaną niedoborem reduktazy sepiapteryny. Ta postać dystonii charakteryzuje szerokie spektrum objawów klinicznych. Pacjenci z łagodnym fenotypem narażeni są na ryzyko niewłaściwej diagnozy – porażenia mózgowego. Osoby o cięższych fenotypach cierpią m.in. na dystonię, zaburzenia mowy, zaburzenia funkcji poznawczych. 47 Przyczyną choroby są mutacje w genie SPR kodującym reduktazę sepiapteryny, enzym zaangażowany w syntezę tetrahydrobiopteryny, kluczowego enzymu szlaku syntezy dopaminy. 48
Dystonie napadowe
Dystonie napadowe charakteryzują się występowaniem epizodów z dystonią lub innymi dyskinezami naprzemiennie z okresami bez zaburzeń neurologicznych. Napadowe dyskinezy z dystonią mogą być prowokowane przez czynniki zewnętrzne. U pacjentów nie obserwuje się oznak procesu neurozwyrodnieniowego. Obecnie znanych jest siedem zespołów dystonii napadowej o różnej etiologii, w tym omówione poniżej dystonie typu 8, 10 i 18.
Dystonia typu 8
Dystonia typu 8 (DYT8) ujawnia się w dzieciństwie lub okresie dojrzewania, 50 a charakterystyczne dla niej napady dystonicze indukowane są często przez alkohol, kofeinę lub stres, rzadziej ruch, zmęczenie lub gorączkę. Trwają one od kilku minut do kilku godzin. Za wystąpienie objawów choroby odpowiedzialne są mutację w genie MR1 kodującym białko wykazujące wysoką homologię do hydrolazy katalizującej konwersję toksycznego metyloglioksalu do bezpiecznego dla organizmu D-mleczanu. Metyloglioksal (związek z grupy aldehydów powstaje na skutek przemian biochemicznych przy udziale specyficznych enzymów bakteryjnych) jest obecny m.in. w kawie, napojach alkoholowych, jest też wytwarzany podczas stresu komórkowego. 51 DYT8 dziedziczy się w sposób autosomalny dominujący.
Dystonia typu 10
Dystonia typu 10 (DYT10) ujawnia się zazwyczaj w dzieciństwie. Charakteryzuje się bardzo częstymi (do 100 razy dziennie) krótkimi atakami ruchów pląsawiczych lub dystonicznych trwającymi od kilku sekund do kilku minut, którym mogą towarzyszyć drgawki. Choroba dziedziczona jest autosomalnie dominująco, spowodowana jest mutacjami w genie PRRT2 kodującym przezbłonowe białko PRRT2 ośrodkowego układu nerwowego, najprawdopodobniej zlokalizowane w synapsach. Mutacje PRRT2 prowadzić mogą do zaburzeń uwalniania neuroprzekaźników, szczególnie w jądrach podstawy, gdzie ulega silnej ekspresji, co sugerowane jest jako patomechanizm DYT10. 52 Spektrum objawów klinicznych spowodowanych mutacjami w genie PRRT2 jest szerokie. Rozpoznanie różnicowe oprócz DYT10 obejmuje: zespół drgawek dziecięcych i choreoatetozy, takie jak napadową dyskinezę wysiłkową oraz napadowe dyskinezy niekinezygeniczne. 53
Dystonia typu 18
Podłoże molekularne dystonii typu 18 (DYT18) stanowią mutacje w genie SLC2A1 kodującym transporter glukozy 1. Choroba dziedziczona jest jako cecha autosomalnie dominująca o szerokim spektrum objawów klinicznych, wynikających z niedoboru białka. Mutacje w tym genie odpowiedzialne są za tzw. zespół niedoboru transportera glukozy GLUT1. Obie choroby traktowane są obecnie jako formy alleliczne i określane jako zespoły niedoboru transportera glukozy GLUT1 typu 1 i typu 2 dla DYT18. Objawy kliniczne DYT18 obejmują dystonię (napady dystoniczne trwają od kilku minut do prawie godziny), 54 opóźnienie psychoruchowe, padaczkę lekooporną, małogłowie, spastyczność, ataksję, drżenie oraz inne dyskinezy. 49 Pacjenci, u których wykryto delecje w obrębie genu, manifestują cięższy fenotyp, natomiast mutacje punktowe wiązane są z mniej poważnymi zaburzeniami ruchowymi i poznawczymi. 55 Ponadto charakterystyczna zmienność objawów powodowana jest przez czynniki środowiskowe, leki lub nawet dietę. Obraz kliniczny choroby może także się zmieniać w ciągu życia. 56, 57
Patomechnizm
Patomechanizm dystonii wydaje się bardziej skomplikowany niż obraz kliniczny choroby. THAP1 (DYT6) oraz TAF1 (DYT3) kodują czynniki transkrypcyjne regulujące ekspresję genów w prążkowiu, których produkty niezbędne są do prawidłowego funkcjonowania m.in. połączeń synaptycznych. 21, 58 Przypuszcza się, że mutacje w CIZ1 (DYT24) mogą zakłócać transkrypcję i regulację genów odgrywających kluczową rolę w rozwoju mózgu lub aktywować cykl komórkowy w pełni zróżnicowanych neuronach np. komórkach Purkiniego. 22 W dystoniach z odpowiedzią na leczenie niskimi dawkami lewodopy (mutacje w genach GCH1, TH, SPR) zaburzony jest szlak syntezy dopaminy. 44, 45, 47 Pośrednio w metabolizm dopaminy zaangażowane jest białko Gαolf (DYT25) pełniące rolę w sprzęganiu receptorów D1 dla dopaminy z cyklazą adenylanową w neuronach prążkowia 26 oraz torsyna A (DYT1) biorąca udział w recyklingu pęcherzyków synaptycznych. 9 Jednak mutacje w genie kodującym TOR1A prowadzą przede wszystkim do zaburzenia struktury otoczki jądrowej oraz siateczki endoplazmatycznej, co bardziej wskazuje na udział białka w dynamice oddziaływań białek cytoszkieletarnych. 11 Ponadto dwa inne białka TUBB4a (DYT4) oraz SGCE (DYT11) wskazują na zaangażowanie w stabilizację cytoszkieletu zarówno komórek mięśniowych, jak i nerwowych. 18, 34 Mutacje w genie kodującym podjednostkę pompy sodowo-potasowej zależnej od ATP (DYT12) oraz kanale chlorkowym aktywowanym jonami wapnia (DYT24) zaburzają funkcjonowanie kanałów jonowych. 39, 24 Nieprawidłowe funkcjonowanie białka PRRT2 (DYT10) oddziałującego m.in. z białkami kompleksu SNARE, biorących udział w fuzji pęcherzyków synaptycznych z błoną komórkową, może skutkować wadliwym przekaźnictwem nerwowym, a tym samym być przyczyną wystąpienia DYT10. 59, 60 W przypadku napadowej dyskinezy niewywołanej ruchem (DYT8) do napadów dystonicznych dochodzi po spożyciu alkoholu i/lub kawy. Podłożem tej choroby są patogenne zmiany w genie MR1 kodującym białko uczestniczące w konwersji metyloglioksalu, toksycznego produktu przemiany m.in. kawy lub alkoholu, do bezpiecznego dla organizmu D-mleczanu. 51 Nieprawidłowe funkcjonowanie białka GLUT1 (DYT18), będącego transporterem glukozy specyficznym dla bariery krew-mózg i erytrocytów, jest przyczyną pojawienia się ruchów dystonicznych. 49
Postępowanie diagnostyczne
<<<Rycina>>>Ze względu na heterogenne podłoże dystonii kluczową rolę w klasyfikacji poszczególnych postaci choroby odgrywa rozpoznanie kliniczne. Diagnozując pacjenta, należy zwrócić szczególną uwagę na wiek zachorowania, lokalizację pierwszych objawów oraz występowanie lub brak rodzinnej historii choroby. W zależności od klinicznego rozpoznania typu dystonii analiza molekularna obejmuje różne geny, a ostateczna weryfikacja molekularna opiera się na wykryciu mutacji w analizowanym genie (rycina).
Spośród dystonii izolowanych analizie molekularnej w pierwszej kolejności poddawany jest gen TOR1A, następnie sugerowana jest diagnostyka molekularna w kierunku DYT6 (gen THAP1). Ruchy dystoniczne obejmujące u pacjenta górne partie mięśni są podstawą do analizy molekularnej genów THAP1, GNAL, CIZ1 oraz ANO3. Jeśli wraz z rozwojem choroby dystonia szyjna przechodzi w postać uogólnioną, dodatkowo pojawiła się charakterystyczna chrypka (2-3 dekada życia) oraz tzw. koński chód, należy rozważyć analizę molekularną TUBB4a. Diagnozując dystonie złożone, ważne jest określenie rytmu, amplitudy, intensywności objawów klinicznych i dodatkowych cech klinicznych. W przypadku dystonii złożonych o charakterze napadów ciągłych z towarzyszącym parkinsonizmem w zależności od obrazu klinicznego choroby oraz typu dziedziczenia powinno się rozważyć analizę genów ATP1A3, PRKRA oraz TAF1. Analiza genu SGCE rekomendowana jest przy współistnieniu dystonii z miokloniami. Dobra odpowiedź na leczenie małymi dawkami lewodopy może wskazywać na mutacje w GCH1, TH oraz SPR. Wśród pacjentów cierpiących na dystonie napadowe istotnym ze względów różnicowania, nie tylko pod względem klinicznym, ale i molekularnym, jest czynnik środowiskowy lub jego brak mogący wywołać napady dystoniczne. W przypadku napadów dystonicznych pojawiających się po spożyciu kawy lub alkoholu należy rozważyć analizę genu MR1, jeśli natomiast napady prowokowane są przez ruch, to w zależności od ich intensywności analizie molekularnej powinien być poddany gen PRRT2 (napady częste >100 na dzień) lub SLC2A1.
Leczenie
Leczenie dystonii jest wyłącznie objawowe i opiera się głównie na stosowaniu różnego rodzaju miorelaksantów oraz na miejscowym blokowaniu/hamowaniu nadmiernej aktywności zajętych mięśni. Domięśniowe podanie toksyny botulinowej hamuje wydzielanie acetylocholiny w miejscu złącza nerwowo-mięśniowego, prowadząc do zmniejszenia napięcia mięśniowego, osłabiając przez to ruchy dystoniczne. Pierwsze efekty widoczne są po kilku dniach od podania leku, utrzymują się przez 3-4 miesięcy, po czym objawy dystonii powracają i potrzebna jest kolejna iniekcja. 5 Wstrzyknięcia toksyny botulinowej mają korzystny efekt również w przypadku dystonii ogniskowych. 61
W postaciach dystonii z odpowiedzią na lewodopę podawanie tego naturalnego, niewchodzącego w skład białka aminokwasu (dawki 20-300 mg) przynosi efekty stałe niezależne od wieku pacjenta i czasu trwania choroby. Leczenie lewodopą w połączeniu z inhibitorem dekarboksylazy aminokwasów aromatycznych stosuje się także w przypadkach dystonii spowodowanej niedoborem reduktazy sepiapteryny, poprawia to funkcjonowanie ruchowe, ale nie wpływa na funkcje poznawcze. Z powodzeniem stosuje się też prekursor serotoniny (5-hydroksytryptofan). 62 Wprowadzenie diety ketogennej oraz dożylnie podawanie glukozy opóźnia rozwój choroby, poprawia stan kliniczny pacjentów z rozpoznaniem DYT18. 63 Objawy mioklonii towarzyszącej DYT11 mogą być częściowo zniesione przez podawanie pacjentom benzodiazepin, leków przeciwpadaczkowych oraz cholinolitycznych.
Opcją terapeutyczną leczenia dystonii, szczególnie postaci uogólnionych, jest głęboka stymulacja mózgu (deep brain stimulation, DBS). Ten rodzaj neurochirurgii czynnościowej polega na wprowadzeniu elektrod, które przez stymulację prądem o wysokiej częstotliwości hamują nadmiernie pobudzone struktury w mózgu. Tradycyjnie celem wszczepienia elektrod w leczeniu dystonii jest część wewnętrzna gałki bladej (Gpi). Ten sposób leczenia jest skuteczny i obarczony niedużą liczbą powikłań, jednak ze względu na inwazyjność jest zarezerwowany do leczenia nasilonych dystonii uogólnionych, takich jak np. DYT1. DBS bywa stosowany w dystoniach ogniskowych, np. w opornym na leczenie toksyną botulinową kręczu karku. 64
Podsumowanie
Genetycznie uwarunkowane postaci dystonii to choroby rzadkie. Dzięki nowym technikom analizy DNA, takim jak eksomowe czy genomowe sekwencjonowanie następnej generacji udało się w krótkim czasie zidentyfikować wiele nowych genów (CIZ1, ANO3, TUBB4a, GNAL), których mutacje są odpowiedzialne za wystąpienie dystonii 22, 24, 18, 25. 15, 16, 21, 22, 23 Nowe technologie umożliwią nie tylko identyfikację nowych genów, ale także opracowanie panelu wariantów znanych genów, co w przyszłości przyczyni się do lepszego zrozumienia patomechanizmów dystonii, a w konsekwencji przy dobrej klasyfikacji umożliwi celowane postępowanie terapeutyczne.
Podziękowania
Praca powstała podczas realizacji projektu badawczego NN 401 135 439.
- 1. Lohmann K, Klein C. Genetics of Dystonia: What’s Known? What’s new? What’s Next? Mov Dis 2013; 28: 899-905.
- 2. Oppenheim H. UbereineeigenartigeKrampfkrankheit des kindlichen und jugendlichen Alters (dysbasialordoticaprogresisiva, Dystonia musculorumdeformans). NeurolCentralbl 1911; 30: 1090-1107.
- 3. EldrigeR .Torsion spasm in Jewish children, and the early history of human genetics. Adv Neurol 1976; 14: 105-114.
- 4. Ichinose H, Ohye T, Takahashi E, at al. Hereditary progressive dystonia with marked diurnal fluctuation caused by mutations in the GTP cyclohydrolase I gene. Nat Genet 1994; 8: 236-242.
- 5. Albanese A, Asmus F, Bhatia KP, at al. EFNS guidelines on diagnosis and treatment of primary dystonias. Eur J Neurol 2011; 18: 5-18.
- 6. Balint B, Bhatia KP. Dystonia: an update on phenomenology, classification, pathogenesis and treatment.Curr Opin Neurol 2014; 27: 468-476.
- 7. Ozelius LJ, Hewett JW, Page CE, at al. The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat Genet 1997; 17: 40-48.
- 8. Bressman SB, de Leon D, Brin MF at al. Idiopathic dystonia among Ashkenazi Jews: evidence for autosomal dominant inheritance. Ann Neurol 1989; 26: 612-620.
- 9. Kakazu Y, Koh JY, Ho KW, et al.Synaptic vesicle recycling is enhanced by torsinA that harbors the DYT1 dystonia mutation. Synapse 2012; 66: 453-464.
- 10. Goodchild RE, Dauer WT. The AAA+ protein torsinA interacts with a conserved domain present in LAP1 and a novel ER protein. J Cell Biol 2005; 168: 855-862.
- 11. Goodchild RE, Kim CE, Dauer WT. Loss of the dystonia-associated protein torsinA selectively disrupts the neuronal nuclear envelope. Neuron 2005; 48: 923-932.
- 12. Gordon KL, Gonzalez-Alegre P. Consequences of the DYT1 mutation on torsinAoligomerization and degradation. Neuroscience 2008; 157: 588-595.
- 13. Cheng FB, Wan XH, Zhang Y, at al.TOR1A sequence variants and the association with early-onset primary dystonia in the Chinese Han population. Parkinsonism Relat Disord 2013; 9: 399-401.
- 14. Calakos N, Patel VD, Gottron M, at al. Functional evidence implicating a novel TOR1A mutation in idiopathic, late-onset focal dystonia. J Med Genet 2010; 47: 646-650.
- 15. Zirn B, Grundmann K, Huppke P, at al. Novel TOR1A mutation p.Arg288Gln in early-onset dystonia (DYT1). J NeurolNeurosurg Psychiatry 2008; 79: 1327-1330.
- 16. Leung JC, Klein C, Friedman J, at al. Novel mutation in the TOR1A (DYT1) gene in atypical early onset dystonia and polymorphisms in dystonia and early onset parkinsonism. Neurogenetics 2001; 3: 133-143.
- 17. Parker N. Hereditary whispering dysphonia. J NeurolNeurosurg Psychiatry 1985; 48: 218-224.
- 18. Hersheson J, Mencacci NE, Davis M. Mutation in the Autoregulatory Domain of β-Tubulin 4a Cause Hereditary Dystonia. Ann Neurol 2013; 73: 546-553.
- 19. Yen TJ, Gay DA, Pachter JS, at al. Autoregulated changes in stability of polyribosome-bound beta-tubulin mRNAs are specified by the first 13 translated nucleotides. Mol Cell Biol 1988; 8: 1224-1235.
- 20. Fuchs T, Gavarini S, Saunders-Pullman R, at al. Mutations in the THAP1 gene are responsible for DYT6 primary torsion dystonia. Nat Genet 2009; 41: 286-288.
- 21. Zhao Y, Xiao J, Gong S, et al. Neural expression of the transcription factor THAP1 during development in rat. Neuroscience 2013; 231: 282-295.
- 22. Xiao J, Uitti RJ, Zhao Y, at al. Mutations in CIZ1 cause adult onset primary cervical dystonia. Ann Neurol 2012; 71: 458-469.
- 23. Klein C, König IR, Lohmann K. Exome sequencing for gene discovery: time to set standard criteria. Ann Neurol 2012; 72: 627-628.
- 24. Charlesworth G, Plagnol V, Holmstrom KM, at al. Mutations in ANO3 cause dominant craniocervical dystonia: ion channel implicated in pathogenesis. Am J Hum Genet 2012; 91: 1041-1050.
- 25. Fuchs T, Saunders-Pullman R, Masuho I, at al. Mutations in GNAL cause primary torsion dystonia. Nat Genet 2012; 45: 88-92.
- 26. Zhuang X, Belluscio L, Hen R. G(olf)alpha mediates dopamine D1 receptor signaling. J Neurosci 2000; 20: RC91.
- 27. Fadool DA, Estey SJ, Ache BW. Evidence that a Gq-protein mediates excitatory odor transduction in lobster olfactory receptor neurons. Chem Senses 1995; 20: 489-498.
- 28. Evidente VG. Zolpidem improves dystonia in “Lubag” or X-linked dystonia-parkinsonism syndrome. Neurology 2002; 58: 662-663.
- 29. Kupke KG, Lee LV, Viterbo GH at al. X-linked recessive torsion dystonia in the Philippines. Am J Med Genet 1990; 36: 237-242.
- 30. Muller U. The monogenic primary dystonias. Brain 2009; 132: 2005-2025.
- 31. Zimprich A, Grabowski M, Asmus F, at al. Mutations in the gene encoding epsilon-sarcoglycan cause myoclonus-dystonia syndrome. Nat Genet 2001; 29: 66-69.
- 32. Roze E, Apartis E, Clot F, at al. Myoclonus-dystonia: clinical and electrophysiologic pattern related to SGCE mutations. Neurology 2008; 70: 1010-1016.
- 33. Chan P, Gonzalez-Maeso J, Ruf F, at al. Epsilon-sarcoglycan immunoreactivity and mRNA expression in mouse brain. J Comp Neurol 2005; 482: 50-73.
- 34. Esapa CT, Waite A, Locke M, at al. SGCE missense mutations that cause myoclonus-dystonia syndrome impair epsilon-sarcoglycan trafficking to the plasma membrane: modulation by ubiquitination and torsinA. Hum Mol Genet 2007; 16: 327-342.
- 35. Guettard E, Portnoi MF, Lohmann-Hedrich K, at al. Myoclonus-dystonia due to maternal uniparental disomy. Arch Neurol 2008; 65: 1380-1385.
- 36. Grabowski M, Zimprich A, Lorenz-Depiereux B, at al. The epsilon-sarcoglycan gene (SGCE), mutated in myoclonus-dystonia syndrome, is maternally imprinted. Eur J Hum Genet 2003; 11: 138-144.
- 37. Brashear A, Deleon D, Bressman SB, at al. Rapid-onset dystonia-parkinsonism in a second family. Neurology 1997; 48: 1066-1069.
- 38. deCarvalhoAguiar P, Sweadner KJ, Penniston JT, at al. Mutations in the Na+/K+ -ATPase alpha3 gene ATP1A3 are associated with rapid-onset dystonia parkinsonism. Neuron 2004; 43: 169-175.
- 39. Rodacker V, Toustrup-Jensen M, Vilsen B. Mutations Phe785Leu and Thr618Met in Na+, K+-ATPase, associated with familial rapid-onset dystonia parkinsonism, interfere with Na+ interaction by distinct mechanisms. J BiolChem 2006; 281: 18539-18548.
- 40. Heinzen EL, Swoboda KJ, Hitomi Y, at al. ATP1A3 cause alternating hemiplegia of childhood. Nat Genet 2012; 44: 1030-1034.
- 41. Camargos S, Scholz S, Simon-Sanchez J, at al. DYT16, a novel young-onset dystonia-parkinsonism disorder: identification of a segregating mutation in the stress-response protein PRKRA. Lancet Neurol 2008; 7: 207-215.
- 42. Patel CV, Handy I, Goldsmith T, at al. PACT, a stress-modulated cellular activator of interferon-induced double-stranded RNA-activated protein kinase, PKR. J Biol Chem 2000; 275: 37993-37998.
- 43. Segawa M, Nomura Y, Nishiyama N. Autosomal dominant guanosine triphosphate cyclohydrolase I deficiency (Segawa disease). Ann Neurol 2003; 54 (Suppl 60: S32-45.
- 44. Ichinose H, Ohye T, Takahashi E, at al. Hereditary progressive dystonia with marked diurnal fluctuation caused by mutations in the GTP cyclohydrolase I gene. Nat Genet 1994; 8: 236-242.
- 45. Bartholomé K, Lüdecke B. Mutations in the tyrosine hydroxylase gene cause various forms of L-dopa-responsive dystonia. Adv Pharmacol 1998; 42: 48-49.
- 46. Zhou QY, Quaife CJ, Palmiter RD. Targeted disruption of the tyrosine hydroxylase gene reveals that catecholamines are required for mouse fetal development. Nature1995; 374: 640-643.
- 47. Bonafe L, Thony B, Penzien JM, at al. Mutations in the sepiapterin reductase gene cause a novel tetrahydrobiopterin-dependent monoamine-neurotransmitter deficiency without hyperphenylalaninemia. Am J Hum Genet 2001; 69: 269-277.
- 48. Steinberger D, Blau N, Goriuonov D, at al. Heterozygous mutation in 5'-untranslated region of sepiapterinreductase gene (SPR) in a patient with dopa-responsive dystonia. Neurogenetics 2004; 5: 187-190.
- 49. Suls A, Dedeken P, Goffin K, at al. Paroxysmal exercise-induced dyskinesia and epilepsy is due to mutations in SLC2A1, encoding the glucose transporter GLUT1. Brain 2008; 131: 1831-1844.
- 50. Bruno MK, Lee HY, Auburger GW, at al. Genotype-phenotype correlation of paroxysmal nonkinesigenic dyskinesia. Neurology 2007; 68: 1782-1789.
- 51. Lee HY, Xu Y, Huang Y, at al. The gene for paroxysmal non-kinesigenic dyskinesia encodes an enzyme in a stress response pathway. Hum Mol Genet 2004; 13: 3161-170.
- 52. Chen WJ, Lin Y, Xiong ZQ, at al. Exome sequencing identifies truncating mutations in PRRT2 that cause paroxysmal kinesigenic dyskinesia. Nat Genet 2011; 43: 1252-1255.
- 53. Lee HY, Huang Y, Bruneau N, at al. Mutations in the novel protein PRRT2 cause paroxysmal kinesigenic dyskinesia with infantile convulsions. Cell Rep 2012; 1: 2-12.
- 54. Weber YG, Storch A, Wuttke TV, at al. GLUT1 mutations are a cause of paroxysmal exertion-induced dyskinesias and induce hemolytic anemia by a cation leak. J Clin Invest 2008; 118: 2157-2168.
- 55. Leen WG, Klepper J, Verbeek MM, at al. Glucose transporter-1 deficiency syndrome: the expanding clinical and genetic spectrum of a treatable disorder. Brain 2010; 133: 655-670.
- 56. Brocnmann K. The expanding phenotype of GLUT-1deficiency syndrome. Brain Dev 2009; 31: 545-553.
- 57. Pascual JM, Wang D, Lecumberri B. GLUT1 deficiency and other glucose transporter diseases. Eur J Endocrinol 2004;150:627-633.
- 58. Herzfeld T, Nolte D, Müller U. Structural and functional analysis of the human TAF1/DYT3 multiple transcript system. Mamm Genome 2007; 18: 787-795.
- 59. Lee HY, Xu Y, Huang Y, at al. The gene for paroxysmal non-kinesigenic dyskinesia encodes an enzyme in a stress response pathway. Hum Mol Genet 2004; 13: 3161-3170.
- 60. Stelzl U, Worm U, Lalowski M, at al. A human protein-protein interaction network: a resource for annotating the proteome. Cell 2005; 122: 957-68.
- 61. Cif L, Valente EM, Hemm S, at al. Deep brain stimulation in myoclonus-dystonia syndrome. Mov Disord 2004; 19: 724-727.
- 62. Steinberger D, Korinthenberg R, Topka H, at al. Dopa-responsive dystonia: mutation analysis of GCH1 and analysis of therapeutic doses of L-dopa. German Dystonia Study Group. Neurology 2000; 55: 1735-1737.
- 63. Weber YG, Storch A, Wuttke TV, at al. GLUT1 mutations are a cause of paroxysmal exertion-induced dyskinesias and induce hemolytic anemia by a cation leak. J Clin Invest 2008; 118: 2157-2168.
- 64. Coubes P, Vayssiere N, El Fertit H, at al. Deep brain stimulation for dystonia. Surgical technique. Stereotact Funct Neurosurg 2002; 78: 183-191.
Następny artykuł:
 Dodaj do ulubionych
Dodaj do ulubionych
