IV edycja Kongresu HPV już 12-13 czerwca! Poznaj najnowsze trendy w profilaktyce, diagnostyce i leczeniu raka szyjki macicy i innych schorzeń związanych z HPV | Sprawdź >
Dystonia typu 8
Dystonia typu 8 (DYT8) ujawnia się w dzieciństwie lub okresie dojrzewania,50 a charakterystyczne dla niej napady dystonicze indukowane są często przez alkohol, kofeinę lub stres, rzadziej ruch, zmęczenie lub gorączkę. Trwają one od kilku minut do kilku godzin. Za wystąpienie objawów choroby odpowiedzialne są mutację w genie MR1 kodującym białko wykazujące wysoką homologię do hydrolazy katalizującej konwersję toksycznego metyloglioksalu do bezpiecznego dla organizmu D-mleczanu. Metyloglioksal (związek z grupy aldehydów powstaje na skutek przemian biochemicznych przy udziale specyficznych enzymów bakteryjnych) jest obecny m.in. w kawie, napojach alkoholowych, jest też wytwarzany podczas stresu komórkowego.51 DYT8 dziedziczy się w sposób autosomalny dominujący.
Dystonia typu 10
Dystonia typu 10 (DYT10) ujawnia się zazwyczaj w dzieciństwie. Charakteryzuje się bardzo częstymi (do 100 razy dziennie) krótkimi atakami ruchów pląsawiczych lub dystonicznych trwającymi od kilku sekund do kilku minut, którym mogą towarzyszyć drgawki. Choroba dziedziczona jest autosomalnie dominująco, spowodowana jest mutacjami w genie PRRT2 kodującym przezbłonowe białko PRRT2 ośrodkowego układu nerwowego, najprawdopodobniej zlokalizowane w synapsach. Mutacje PRRT2 prowadzić mogą do zaburzeń uwalniania neuroprzekaźników, szczególnie w jądrach podstawy, gdzie ulega silnej ekspresji, co sugerowane jest jako patomechanizm DYT10.52 Spektrum objawów klinicznych spowodowanych mutacjami w genie PRRT2 jest szerokie. Rozpoznanie różnicowe oprócz DYT10 obejmuje: zespół drgawek dziecięcych i choreoatetozy, takie jak napadową dyskinezę wysiłkową oraz napadowe dyskinezy niekinezygeniczne.53
Dystonia typu 18
Podłoże molekularne dystonii typu 18 (DYT18) stanowią mutacje w genie SLC2A1 kodującym transporter glukozy 1. Choroba dziedziczona jest jako cecha autosomalnie dominująca o szerokim spektrum objawów klinicznych, wynikających z niedoboru białka. Mutacje w tym genie odpowiedzialne są za tzw. zespół niedoboru transportera glukozy GLUT1. Obie choroby traktowane są obecnie jako formy alleliczne i określane jako zespoły niedoboru transportera glukozy GLUT1 typu 1 i typu 2 dla DYT18. Objawy kliniczne DYT18 obejmują dystonię (napady dystoniczne trwają od kilku minut do prawie godziny),54 opóźnienie psychoruchowe, padaczkę lekooporną, małogłowie, spastyczność, ataksję, drżenie oraz inne dyskinezy.49 Pacjenci, u których wykryto delecje w obrębie genu, manifestują cięższy fenotyp, natomiast mutacje punktowe wiązane są z mniej poważnymi zaburzeniami ruchowymi i poznawczymi.55 Ponadto charakterystyczna zmienność objawów powodowana jest przez czynniki środowiskowe, leki lub nawet dietę. Obraz kliniczny choroby może także się zmieniać w ciągu życia.56,57
Patomechnizm
Patomechanizm dystonii wydaje się bardziej skomplikowany niż obraz kliniczny choroby. THAP1 (DYT6) oraz TAF1 (DYT3) kodują czynniki transkrypcyjne regulujące ekspresję genów w prążkowiu, których produkty niezbędne są do prawidłowego funkcjonowania m.in. połączeń synaptycznych.21,58 Przypuszcza się, że mutacje w CIZ1 (DYT24) mogą zakłócać transkrypcję i regulację genów odgrywających kluczową rolę w rozwoju mózgu lub aktywować cykl komórkowy w pełni zróżnicowanych neuronach np. komórkach Purkiniego.22 W dystoniach z odpowiedzią na leczenie niskimi dawkami lewodopy (mutacje w genach GCH1, TH, SPR) zaburzony jest szlak syntezy dopaminy.44,45,47 Pośrednio w metabolizm dopaminy zaangażowane jest białko Gαolf (DYT25) pełniące rolę w sprzęganiu receptorów D1 dla dopaminy z cyklazą adenylanową w neuronach prążkowia26 oraz torsyna A (DYT1) biorąca udział w recyklingu pęcherzyków synaptycznych.9 Jednak mutacje w genie kodującym TOR1A prowadzą przede wszystkim do zaburzenia struktury otoczki jądrowej oraz siateczki endoplazmatycznej, co bardziej wskazuje na udział białka w dynamice oddziaływań białek cytoszkieletarnych.11 Ponadto dwa inne białka TUBB4a (DYT4) oraz SGCE (DYT11) wskazują na zaangażowanie w stabilizację cytoszkieletu zarówno komórek mięśniowych, jak i nerwowych.18,34 Mutacje w genie kodującym podjednostkę pompy sodowo-potasowej zależnej od ATP (DYT12) oraz kanale chlorkowym aktywowanym jonami wapnia (DYT24) zaburzają funkcjonowanie kanałów jonowych.39,24 Nieprawidłowe funkcjonowanie białka PRRT2 (DYT10) oddziałującego m.in. z białkami kompleksu SNARE, biorących udział w fuzji pęcherzyków synaptycznych z błoną komórkową, może skutkować wadliwym przekaźnictwem nerwowym, a tym samym być przyczyną wystąpienia DYT10.59,60 W przypadku napadowej dyskinezy niewywołanej ruchem (DYT8) do napadów dystonicznych dochodzi po spożyciu alkoholu i/lub kawy. Podłożem tej choroby są patogenne zmiany w genie MR1 kodującym białko uczestniczące w konwersji metyloglioksalu, toksycznego produktu przemiany m.in. kawy lub alkoholu, do bezpiecznego dla organizmu D-mleczanu.51 Nieprawidłowe funkcjonowanie białka GLUT1 (DYT18), będącego transporterem glukozy specyficznym dla bariery krew-mózg i erytrocytów, jest przyczyną pojawienia się ruchów dystonicznych.49
Postępowanie diagnostyczne
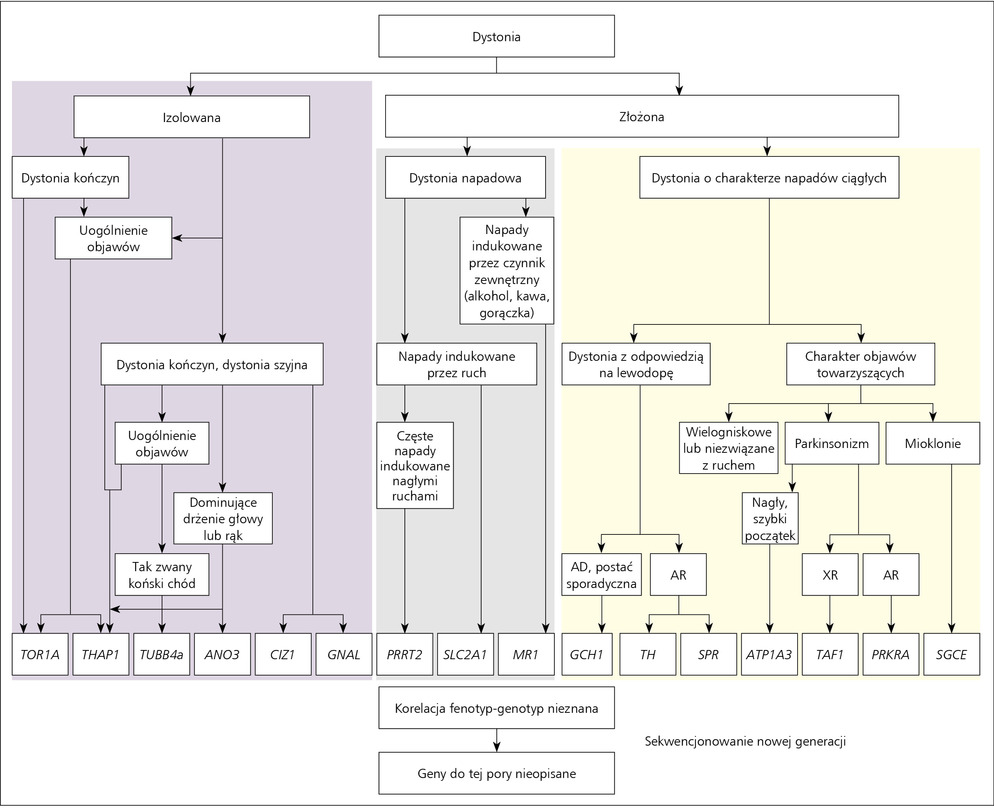
Rycina. Schemat postępowania diagnostycznego u chorych z klinicznym rozpoznaniem dystonii.
Rycina Ze względu na heterogenne podłoże dystonii kluczową rolę w klasyfikacji poszczególnych postaci choroby odgrywa rozpoznanie kliniczne. Diagnozując pacjenta, należy zwrócić szczególną uwagę na wiek zachorowania, lokalizację pierwszych objawów oraz występowanie lub brak rodzinnej historii choroby. W zależności od klinicznego rozpoznania typu dystonii analiza molekularna obejmuje różne geny, a ostateczna weryfikacja molekularna opiera się na wykryciu mutacji w analizowanym genie (rycina).
Spośród dystonii izolowanych analizie molekularnej w pierwszej kolejności poddawany jest gen TOR1A, następnie sugerowana jest diagnostyka molekularna w kierunku DYT6 (gen THAP1). Ruchy dystoniczne obejmujące u pacjenta górne partie mięśni są podstawą do analizy molekularnej genów THAP1, GNAL, CIZ1 oraz ANO3. Jeśli wraz z rozwojem choroby dystonia szyjna przechodzi w postać uogólnioną, dodatkowo pojawiła się charakterystyczna chrypka (2-3 dekada życia) oraz tzw. koński chód, należy rozważyć analizę molekularną TUBB4a. Diagnozując dystonie złożone, ważne jest określenie rytmu, amplitudy, intensywności objawów klinicznych i dodatkowych cech klinicznych. W przypadku dystonii złożonych o charakterze napadów ciągłych z towarzyszącym parkinsonizmem w zależności od obrazu klinicznego choroby oraz typu dziedziczenia powinno się rozważyć analizę genów ATP1A3, PRKRA oraz TAF1. Analiza genu SGCE rekomendowana jest przy współistnieniu dystonii z miokloniami. Dobra odpowiedź na leczenie małymi dawkami lewodopy może wskazywać na mutacje w GCH1, TH oraz SPR. Wśród pacjentów cierpiących na dystonie napadowe istotnym ze względów różnicowania, nie tylko pod względem klinicznym, ale i molekularnym, jest czynnik środowiskowy lub jego brak mogący wywołać napady dystoniczne. W przypadku napadów dystonicznych pojawiających się po spożyciu kawy lub alkoholu należy rozważyć analizę genu MR1, jeśli natomiast napady prowokowane są przez ruch, to w zależności od ich intensywności analizie molekularnej powinien być poddany gen PRRT2 (napady częste >100 na dzień) lub SLC2A1.
Leczenie
Leczenie dystonii jest wyłącznie objawowe i opiera się głównie na stosowaniu różnego rodzaju miorelaksantów oraz na miejscowym blokowaniu/hamowaniu nadmiernej aktywności zajętych mięśni. Domięśniowe podanie toksyny botulinowej hamuje wydzielanie acetylocholiny w miejscu złącza nerwowo-mięśniowego, prowadząc do zmniejszenia napięcia mięśniowego, osłabiając przez to ruchy dystoniczne. Pierwsze efekty widoczne są po kilku dniach od podania leku, utrzymują się przez 3-4 miesięcy, po czym objawy dystonii powracają i potrzebna jest kolejna iniekcja.5 Wstrzyknięcia toksyny botulinowej mają korzystny efekt również w przypadku dystonii ogniskowych.61
W postaciach dystonii z odpowiedzią na lewodopę podawanie tego naturalnego, niewchodzącego w skład białka aminokwasu (dawki 20-300 mg) przynosi efekty stałe niezależne od wieku pacjenta i czasu trwania choroby. Leczenie lewodopą w połączeniu z inhibitorem dekarboksylazy aminokwasów aromatycznych stosuje się także w przypadkach dystonii spowodowanej niedoborem reduktazy sepiapteryny, poprawia to funkcjonowanie ruchowe, ale nie wpływa na funkcje poznawcze. Z powodzeniem stosuje się też prekursor serotoniny (5-hydroksytryptofan).62 Wprowadzenie diety ketogennej oraz dożylnie podawanie glukozy opóźnia rozwój choroby, poprawia stan kliniczny pacjentów z rozpoznaniem DYT18.63 Objawy mioklonii towarzyszącej DYT11 mogą być częściowo zniesione przez podawanie pacjentom benzodiazepin, leków przeciwpadaczkowych oraz cholinolitycznych.
Opcją terapeutyczną leczenia dystonii, szczególnie postaci uogólnionych, jest głęboka stymulacja mózgu (deep brain stimulation, DBS). Ten rodzaj neurochirurgii czynnościowej polega na wprowadzeniu elektrod, które przez stymulację prądem o wysokiej częstotliwości hamują nadmiernie pobudzone struktury w mózgu. Tradycyjnie celem wszczepienia elektrod w leczeniu dystonii jest część wewnętrzna gałki bladej (Gpi). Ten sposób leczenia jest skuteczny i obarczony niedużą liczbą powikłań, jednak ze względu na inwazyjność jest zarezerwowany do leczenia nasilonych dystonii uogólnionych, takich jak np. DYT1. DBS bywa stosowany w dystoniach ogniskowych, np. w opornym na leczenie toksyną botulinową kręczu karku.64
Podsumowanie
Genetycznie uwarunkowane postaci dystonii to choroby rzadkie. Dzięki nowym technikom analizy DNA, takim jak eksomowe czy genomowe sekwencjonowanie następnej generacji udało się w krótkim czasie zidentyfikować wiele nowych genów (CIZ1, ANO3, TUBB4a, GNAL), których mutacje są odpowiedzialne za wystąpienie dystonii 22, 24, 18, 25.15,16,21-23 Nowe technologie umożliwią nie tylko identyfikację nowych genów, ale także opracowanie panelu wariantów znanych genów, co w przyszłości przyczyni się do lepszego zrozumienia patomechanizmów dystonii, a w konsekwencji przy dobrej klasyfikacji umożliwi celowane postępowanie terapeutyczne.
Podziękowania
Praca powstała podczas realizacji projektu badawczego NN 401 135 439.