Wyróżnia się trzy główne postaci choroby: dystrofię neuroaksonalną Seitelbergera (postać wczesnodziecięca), atypową dystrofię neuroaksonalną (ANAD – atypical neuroaxonal dystrophy) i zespół dystonia-parkinsonizm (PARK14).1-4 Postać wczesnodziecięca przebiega z zahamowaniem rozwoju psychoruchowego oraz napadami padaczkowymi, a śmierć następuje w ciągu kilku lat. W ANAD objawy pojawiają się nieco później. Najczęściej obserwuje się dystonię, zaburzenia psychiatryczne oraz napady padaczkowe. Zespół dystonia-parkinsonizm rozpoczyna się w wieku 20-30 lat i charakteryzuje dobrą odpowiedzią na lewodopę. MR mózgu wykazuje odkładanie się żelaza w gałkach bladych, zaniki móżdżku i uszkodzenie istoty białej. Badanie genetyczne potwierdza rozpoznanie. Leczenie zespołu jest wyłącznie objawowe.1,4,6
Neurodegeneracja związana z białkiem błony mitochondrialnej
Za neurodegenerację związaną z białkiem błony mitochondrialnej (MPAN – mitochondrial membrane protein-associated neurodegeneration) odpowiada gen C19orf12, który znajduje się na chromosomie 19. Objawy pojawiają się stopniowo po 9 r.ż. Często obserwuje się dystonie, parkinsonizm, zaburzenia zachowania i objawy piramidowe. Ponadto występują polineuropatia aksonalna oraz zanik nerwów wzrokowych. MR mózgu wykazuje zmiany hipodensyjne w obrębie gałek bladych oraz istoty czarnej w sekwencjach T2, natomiast nie opisuje się klasycznego oka tygrysa w tym badaniu. Do potwierdzenia rozpoznania służy badanie genetyczne. Leczenie jest tylko objawowe.1-4,8
Choroby związane z gromadzeniem manganu
Bardzo rzadka choroba genetyczna związana z gromadzeniem manganu spowodowana jest mutacją w genie SLC30A10, który koduje transporter manganu. Skutkuje to odkładaniem się manganu głównie w jądrach podstawy i w móżdżku.11,12
Pierwsze objawy pod postacią dystonii uogólnionej zwykle pojawiają się w 1 dekadzie życia. Występują również zaburzenia mowy o typie dyzartrii, zespół parkinsonowski, neuropatia ruchowa oraz zaburzenia psychiatryczne. Charakterystycznym objawem jest marskość wątroby. W preparatach z biopsji wątroby stwierdza się podwyższone stężenie manganu, a w badaniach laboratoryjnych – policytemię oraz obniżone stężenie ferrytyny i żelaza w surowicy. MR głowy w sekwencji T1 wykazuje hiperintensywne zmiany zlokalizowane głównie w gałkach bladych, a także w prążkowiu, móżdżku i istocie białej.11,12

Rycina 2. MR głowy. Hiperintensywne zmiany w obrazach T1-zależnych w gałkach bladych związane z gromadzeniem się manganu u pacjentki nadużywającej efedronu
W chorobie związanej z gromadzeniem manganu stosuje się leczenie chelatujące z wykorzystaniem soli wapniowo-disodowej kwasu wersenowego, prowadzące do wzmożonego wydalania manganu. W większości przypadków następuje poprawa kliniczna. Dodatkowo prowadzi się suplementację preparatów żelaza, ponieważ żelazo stabilizuje stężenie manganu w surowicy.11,12
Przyczyną odkładania się manganu w ośrodkowym układzie nerwowym (OUN) może być również nadużywanie efedronu (ryc. 2). Efedron (metylokatynon) jest inhibitorem wychwytu zwrotnego noradrenaliny oraz dopaminy i działa jako psychostymulant. Dodatek stosowany do sporządzania roztworu do iniekcji dożylnych zawiera nadmanganian potasu. W tak uzyskanym roztworze znajduje się duża ilość manganu, który odkłada się w OUN, głównie w jądrach podkorowych. W diagnostyce różnicowej należy uwzględnić nabyte zwyrodnienie wątrobowo-soczewkowe w schyłkowej niewydolności wątroby.5
Choroba Wilsona
Choroba Wilsona to rzadka choroba dziedziczona autosomalnie recesywnie, w której dochodzi do zaburzenia metabolizmu miedzi i jej odkładania się w różnych narządach, głównie w wątrobie i mózgu. Najczęściej u chorych obserwuje się objawy uszkodzenia wątroby, objawy neurologiczne i psychiatryczne.13-15 Przyczyną zaburzeń metabolizmu miedzi są mutacje w genie ATP7B zlokalizowanym na chromosomie 13.14 Gen ten koduje białko ATP-azę, która pośredniczy we wbudowywaniu miedzi w apoceruloplazminę. Na skutek nieprawidłowego funkcjonowania ATP-azy miedź gromadzi się w hepatocytach, uszkadza je, następnie jest uwalniana do krwiobiegu i powoduje uszkodzenie kolejnych narządów: mózgu, rogówki, nerek oraz serca.14
Objawy kliniczne
Pierwsze symptomy choroby obserwuje się najczęściej między 10 a 40 r.ż. Obraz kliniczny jest zróżnicowany: występują objawy uszkodzenia wątroby, neurologiczne oraz psychiatryczne,13-15 przy czym objawy neurologiczne zwykle rozwijają się później niż objawy uszkodzenia wątroby. Te ostatnie mogą się ujawniać jako wzrost aktywności enzymów wątrobowych, bezobjawowe powiększenie wątroby i śledziony. Występują także ostre i przewlekłe zapalenie wątroby oraz marskość. Wśród objawów neurologicznych dominują zaburzenia mowy, drżenie (pozycyjne, zamiarowe, spoczynkowe), dystonia i zaburzenia chodu.
Choroba Wilsona daje dwa charakterystyczne objawy: drżenie przypominające bicie skrzydłami, które dotyczy proksymalnych części kończyn górnych,16 oraz pierścień Kaysera-Fleischera, będący efektem odkładania się miedzi w przednich warstwach rogówki oka na błonie Descemeta.13 Poza opisanymi najczęściej występującymi objawami można zaobserwować również zaburzenia hematologiczne, endokrynologiczne, kostno-stawowe, nefrologiczne i kardiologiczne.13,14
Dla celów praktycznych została opracowana skala do oceny stanu neurologicznego pacjentów z chorobą Wilsona, pozwalająca ocenić nasilenie objawów neurologicznych i stan funkcjonalny.17
Diagnostyka
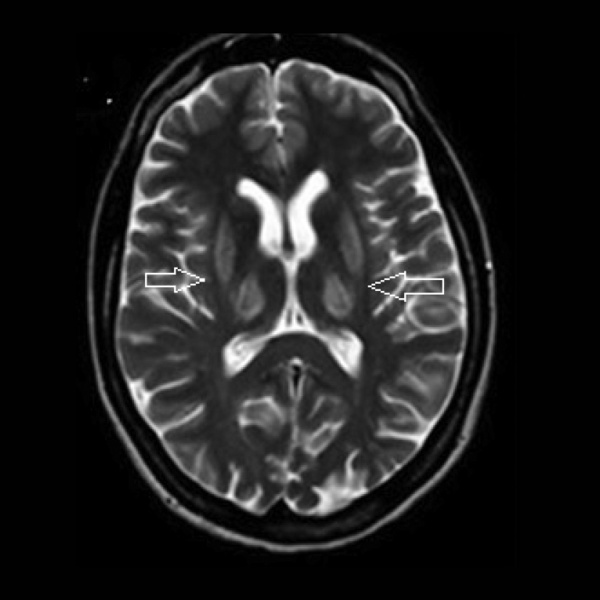
Rycina 3. MR głowy. Hiperintensywne zmiany w obrazach T2-zależnych w skorupach i we wzgórzach u pacjentki z chorobą Wilsona
Rozpoznanie opiera się na kombinacji objawów klinicznych, wyników badań laboratoryjnych i neuroobrazowych oraz identyfikacji mutacji w genie ATP7B.13-15 W przypadku podejrzenia choroby Wilsona należy wykonać badania metabolizmu miedzi: oznaczenie stężenia ceruloplazminy (zwykle obniżone o więcej niż 50% dolnej granicy normy) i miedzi całkowitej w surowicy (na ogół obniżone) oraz badanie wydalania miedzi w dobowej zbiórce moczu (najczęściej podwyższone). U osób chorych wydalanie miedzi w dobowej zbiórce moczu wzrasta do ponad 100 g/24 h.13 W niektórych przypadkach stosuje się również test z wbudowywaniem miedzi radioaktywnej do ceruloplazminy.15